Nature | 郑州大学校长李蓬院士联手北大/清华团队揭示肝脏脂质代谢新机制,内质网膜蛋白CLCC1指挥磷脂跨膜运输,维持全身代谢平衡
脂质的合成、储存和动员对细胞和系统稳态至关重要。内质网(ER)是细胞脂质合成与分泌的核心枢纽,特别是在代谢活跃的肝细胞和肠道上皮细胞中。脂蛋白的组装需磷脂跨 ER 膜转运至管腔载脂蛋白 B(APOB),但该过程的调控机制未明确。

2026年02月25日,北京大学陈晓伟教授、郑州大学校长李蓬院士、清华大学徐俐副研究员及北京大学郭强研究员作为共同通讯作者在《Nature》期刊发表研究论文:“CLCC1 Governs ER Bilayer Equilibration to Maintain LipidHomeostasis”,北京大学吴凌志博士、清华大学王剑琴博士和北京大学王雅伟博士为共同第一作者。
研究聚焦ER磷脂跨双层平衡的调控机制,鉴定出 ER 蛋白CLCC1为脂质稳态的核心调控因子,揭示了其与 TMEM41B 协同调控磷脂翻转、脂蛋白生物发生及肝脏代谢稳态的分子与生理机制,研究从细胞、动物、人类遗传多态性三个层面展开,结合多种生物化学、细胞生物学和影像学技术,明确了 CLCC1 的功能与作用机制,为代谢功能障碍相关肝脂肪性肝炎(MASH)等疾病的研究提供了新靶点。
一、研究背景:ER 脂质代谢的关键科学问题
1. 磷脂翻转酶TMEM41B是 ER 磷脂跨双层平衡的关键因子,肝脏 TMEM41B 缺失会阻断极低密度脂蛋白(VLDL)生物发生,诱导脂质过度合成并加速代谢功能障碍相关肝脂肪性肝炎(MASH)发病,但其调控因子未知。
2. 脂代谢紊乱是心代谢疾病的主要危险因素,而 ER 膜磷脂失衡与脂滴异常形成的关联尚未被系统解析。
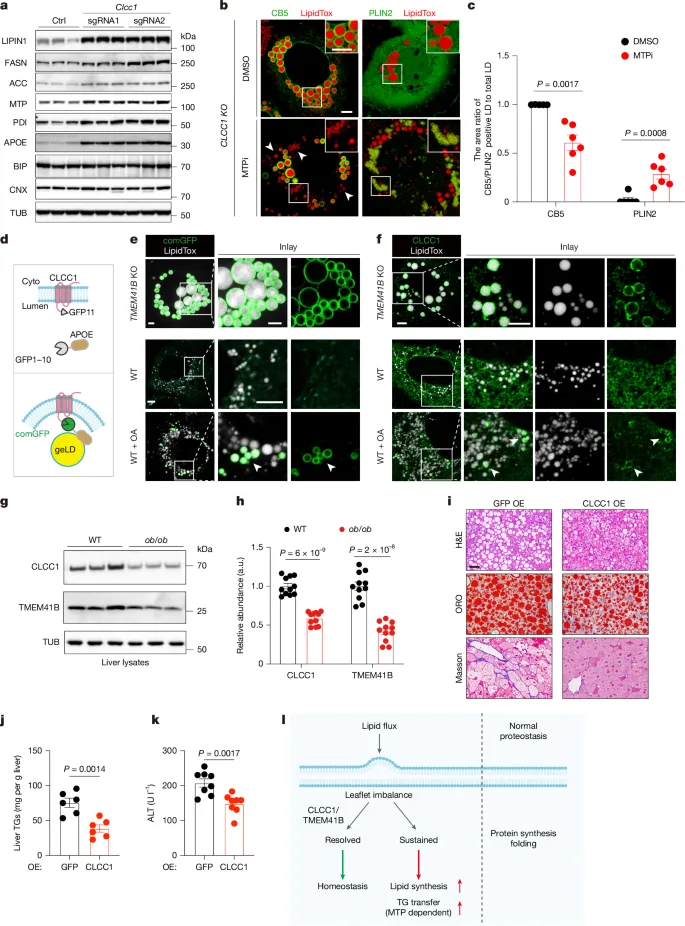
二、TMEM41B 缺失诱导巨型内质网包裹脂滴(geLDs) 形成
1. 形态学特征:通过 TEM 和 cryo-ET 发现,TMEM41B 敲除小鼠肝细胞中形成直径约1 μm的 geLDs(野生型脂滴为胞质分布,平均 ER 管腔宽度 50 nm),geLDs 由无核糖体的光滑 ER 双层膜包裹,膜磷脂存在胞质 / 管腔叶失衡。
2. 分子特征:geLDs 缺乏经典胞质脂滴(cLDs)标志物PLIN2,富集 ER 膜蛋白(如 SIGMAR1、细胞色素 b5),且缺失 VLDL 核心结构蛋白 APOB,仅含载脂蛋白 APOE。
3. 磷脂失衡验证:薄层层析(TLC)显示,geLDs 的磷脂含量仅为同尺寸 cLDs 的1.5N(理论匹配膜为 3N),证实 ER 膜管腔侧存在磷脂短缺。
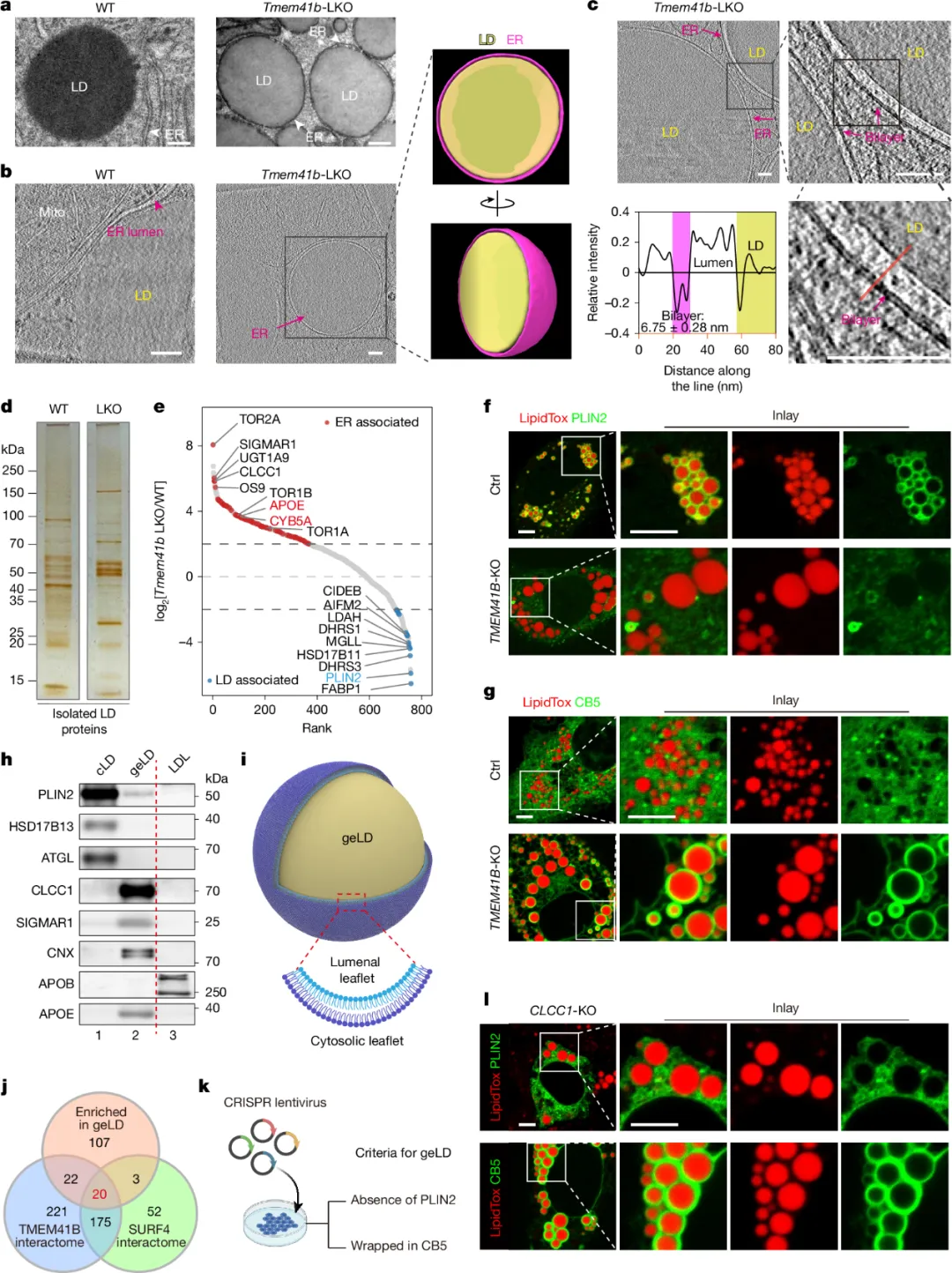
三、CLCC1 的鉴定:geLDs 富集的 ER 膜蛋白,与 TMEM41B 协同作用
1. 筛选与鉴定:通过 geLDs 富集蛋白质组、TMEM41B 互作组和 SURF4 互作组的交集分析,鉴定出CLCC1为核心候选蛋白,其在 geLDs 周围的 ER 膜中高度富集。
2. 相互作用验证
(1)串联亲和纯化和免疫共沉淀证实 CLCC1 与 TMEM41B 形成内源性蛋白复合物;
(2)肝脏 CLCC1 敲除会导致 TMEM41B 蛋白水平下调(转录水平不变),证实 CLCC1 对 TMEM41B 的蛋白稳定性至关重要。
3. 人类遗传关联:基于 GLGC 数据库的 GWAS 分析显示,人类CLCC1 基因 rs149700491 位点与血浆血脂水平显著相关,其中与低密度脂蛋白胆固醇(LDL-C)的关联最显著,该位点次要等位基因频率在欧亚人群中均低于 0.01。
四、CLCC1 缺失的细胞与动物表型:脂质代谢紊乱与 MASH 加速发病
1. 细胞表型:CRISPR 敲除 CLCC1 会重现 TMEM41B 缺失的表型,即 geLDs 形成、磷脂跨膜翻转受损、APOB 脂化失败,VLDL 合成受阻。
2. 小鼠体内表型(AAV 介导的肝脏 CLCC1 急性敲除)
检测指标 | 敲除 CLCC1 小鼠表型 |
空腹血浆甘油三酯 | 降至近零 |
血浆 VLDL/LDL | 完全耗竭 |
血浆总胆固醇 | 显著降低 |
肝脏重量 / 体重比 | 增加近 1 倍 |
肝脏甘油三酯含量 | 显著升高 |
血浆 ALT(肝损伤指标) | 大幅升高 |
3.病理表型:肝脏 CLCC1 敲除小鼠在无高脂饮食诱导的 4 周内快速发展为 MASH,表现为肝细胞气球样变、肝纤维化、免疫细胞浸润,肝脏密度降低可浮于水面,上述表型可通过回补 CLCC1 完全挽救。
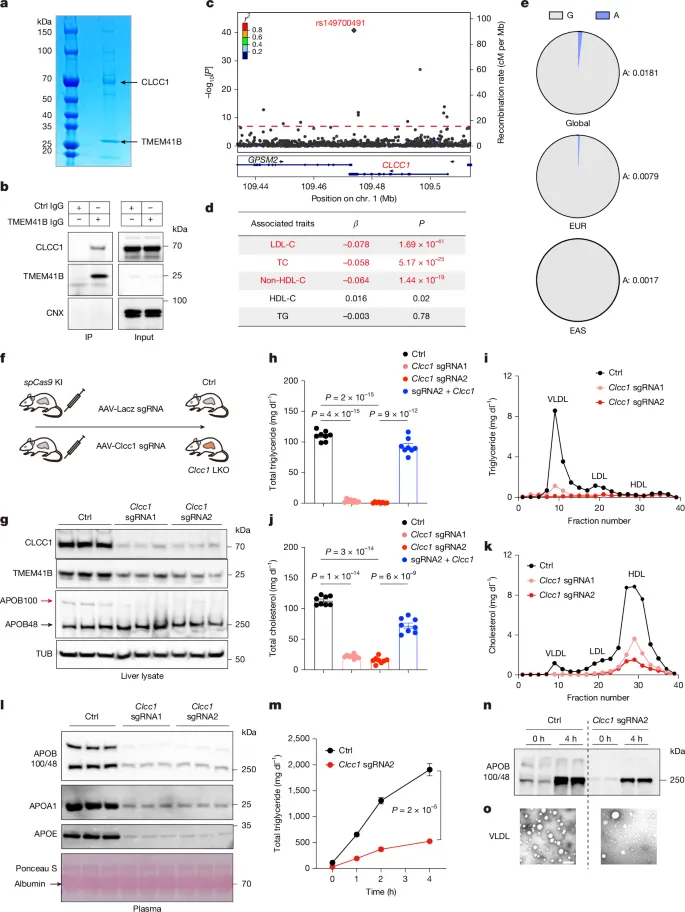
五、CLCC1 的分子机制:调控 TMEM41B 介导的 ER 磷脂跨双层平衡
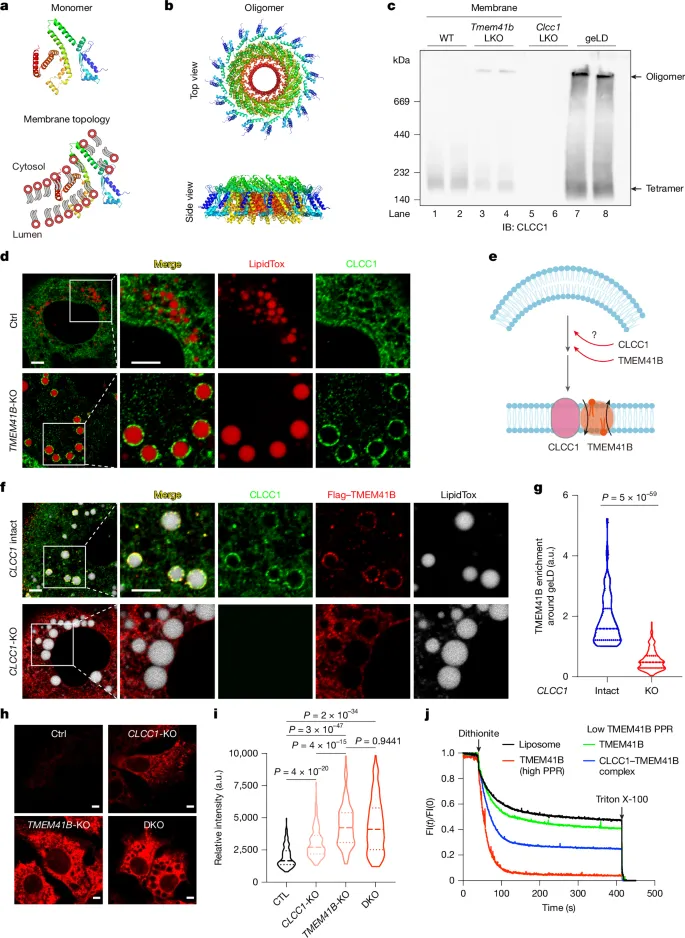
1. CLCC1 的结构与定位:AlphaFold3 预测 CLCC1 形成环状寡聚体,其管腔域是寡聚化和定位于 geLDs 周围失衡 ER 膜的关键;缺失管腔域的 CLCC1 无法寡聚化,也不能挽救 geLDs 形成。
2. 招募 TMEM41B 至失衡 ER 膜:CLCC1 是 TMEM41B 定位于磷脂失衡 ER 膜的必要条件,CLCC1 敲除后,外源性 TMEM41B 无法被招募至 geLDs 周围,磷脂翻转功能丧失。
3. 调控磷脂翻转活性
(1)细胞水平:CLCC1 敲除导致 ER 胞质侧磷脂(PC)蓄积,磷脂翻转效率下降(低于 TMEM41B 敲除但显著高于野生型);
(2)体外实验:CLCC1 本身无磷脂翻转酶活性,但与 TMEM41B 形成复合物后,可使 TMEM41B 的磷脂翻转活性显著增强,且无脂双层渗漏。
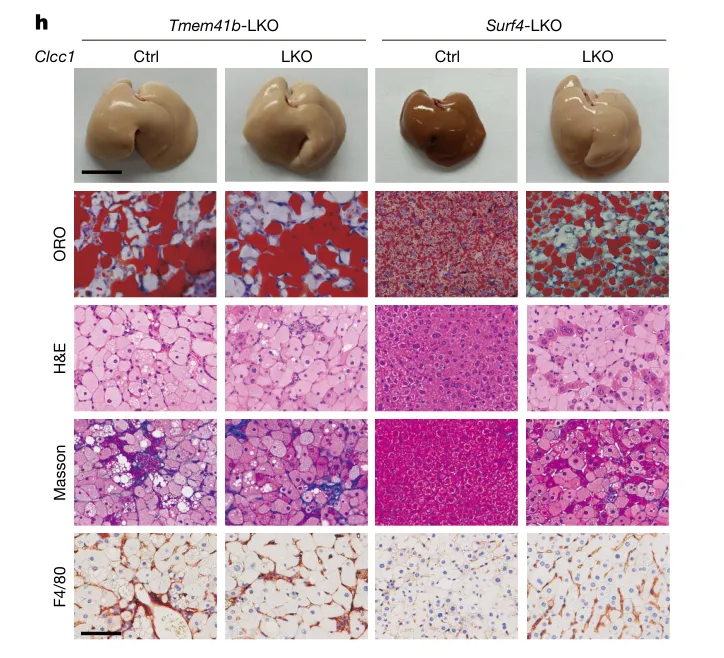
4. 遗传上位性分析:CLCC1 与 TMEM41B 双敲除小鼠的肝病理表型未比 TMEM41B 单敲除加重,证实二者作用于同一磷脂翻转通路;而 CLCC1 与 SURF4 双敲除表型与 CLCC1 单敲除一致,证实 CLCC1 作用于 SURF4 介导的脂蛋白转运上游。
六、CLCC1 拮抗脂质诱导的 ER 应激,缓解肥胖相关肝损伤
1. CLCC1 与脂质应激的关联:油酸处理的野生型肝细胞中,CLCC1 会重新定位至脂滴周围,形成短暂的磷脂失衡 ER 膜,提示 CLCC1 参与生理状态下的脂质应激响应。
2. 肥胖模型中的表达变化:高脂饮食(HFD)和 ob/ob 肥胖小鼠的肝脏中,CLCC1 和 TMEM41B 蛋白水平均下调约 50%,与肝脏脂质蓄积和 ER 膜失衡相关。
3. 过表达的治疗效果:AAV 介导的肝脏 CLCC1 过表达可显著缓解 ob/ob 小鼠的肝脏脂质蓄积(甘油三酯降低)、肝损伤(血浆 ALT/AST 下调)和肝纤维化,同时上调 TMEM41B 蛋白水平,抑制脂合成酶(如 LIPIN1)的表达。
七、研究意义与结论
1. 机制意义:首次阐明了 CLCC1 作为 TMEM41B 的调控因子,通过识别 ER 膜磷脂双层失衡、招募并增强 TMEM41B 的磷脂翻转活性,维持 ER 膜的动态平衡,填补了 ER 磷脂跨双层转运调控机制的空白。
2. 生理意义:证实 ER 磷脂翻转是脂蛋白生物发生、细胞脂质分配和全身脂质稳态的核心环节,CLCC1 是脂代谢稳态的关键调控因子。
3. 临床意义:发现 CLCC1 基因遗传变异与人类血浆血脂水平相关,肝脏 CLCC1 过表达可缓解肥胖相关肝损伤,为 MASH、高胆固醇血症等脂代谢相关疾病提供了新的潜在治疗靶点。
4. 延伸意义:ER 磷脂翻转参与自噬、病毒感染等多种生物学过程,本研究为解析 CLCC1 在其他生理病理过程中的功能奠定了基础。
本文原创仅指编译原创,文献内容与图片版权归原著所有,文献解读仅用于学术分享,如有侵权请与后台联系
关注公众号了解更多医院招聘、科研快讯、文献解读和期刊选择相关内容

本公司致力于课题设计、项目申报、组学分析、细胞实验、动物实验、论文指导等多项科研服务,扫描下方二维码可免费咨询


