郑州大学姜辉/龚军芳/宋毛平教授 PS&T 封面:给镍催化剂装上“氟引擎”实现乙烯基弹性体与极性功能化聚烯烃材料精准构筑
- 2026-05-23 22:25:20

聚烯烃材料是当前世界上产量最高、应用最广的高分子材料之一。其中,聚乙烯弹性体和极性功能化聚烯烃是十分热门的高性能聚烯烃材料。然而,这两种材料的直接合成仍面临显著挑战。因此,开发更高效的聚合催化剂,以及更高性能的聚烯烃材料是学术界研究的热点问题。郑州大学化学学院姜辉/龚军芳教授在高性能聚烯烃催化剂和材料研究方面取得了一系列成果(Chem. Eng. J.2026, 529, 172925; Chin. J. Chem.2026, 44, 15; Eur. Polym. J.2026, 246, 114560; Polym. Sci. Technol. 2026. 10.1021/polymscitech.5c00139; Polymer2026, 342, 129317; Sci. China Chem.2025, 68, 714; Molecules2025, 30, 1530; Eur. J. Org. Chem. 2025, e202500471; Polymers 2024, 16, 578; Inorg. Chem. 2023, 62, 5105; Chin. Chem. Lett. 2023, 34, 107918; 高分子学报2023, 54,186; Eur. J. Inorg. Chem. 2023, 26, e202200725.)。
以廉价的乙烯分子为单一原料,直接合成高性能聚乙烯弹性体,是该领域极具应用潜力的发展方向。同时,在非极性的聚烯烃上高效、可控地引入极性官能团,是高分子合成中的一项终极挑战。在已发展的催化剂体系中,Brookhart型α-二亚胺Ni/Pd催化剂凭借其独特的“链行走”机制与优异的共聚性能而备受关注,其不仅能合成支化结构可调的聚乙烯,还可实现乙烯与极性单体的共聚,展现出兼顾合成上述两种材料的潜力。
近年来,在催化剂设计中引入氟原子已成为一种有效的调控策略。然而,如何在单一催化体系中兼顾聚乙烯弹性体的高效合成与极性聚烯烃的可控制备,仍是当前亟需解决的关键难题。针对这一挑战,郑州大学姜辉/龚军芳/宋毛平教授团队近期设计并合成了一系列新型非对称α-二亚胺镍(II)配合物(图1)。通过系统的催化性能评估与聚合物结构解析,研究团队深入探讨了位阻效应和氟效应在催化活性、聚合物分子量及极性单体共聚插入率等方面的协同调控机制。研究结果表明,N-芳基邻位的位阻基团与氟取代之间存在显著的协同作用:一方面促进了高分子量聚乙烯弹性体的形成,另一方面有效提升了乙烯与极性单体的共聚效率,实现了两类关键性能的同步优化。该工作不仅为深入理解α-二亚胺镍催化剂的结构—性能关系提供了重要实验依据,也为新一代高性能聚乙烯弹性体及极性聚烯烃材料的分子设计与开发奠定了坚实基础。
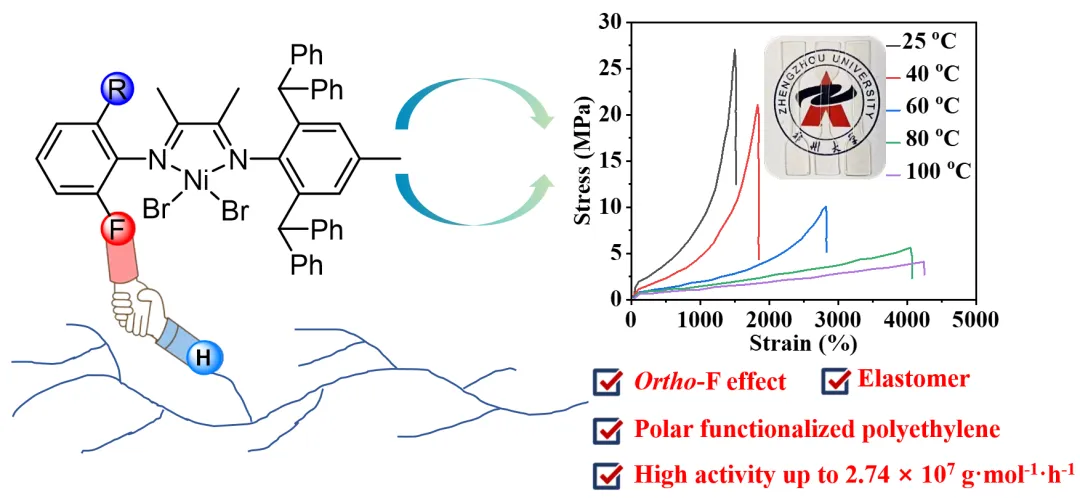
图1. 氟与位阻协同制备高性能聚烯烃
催化剂的合成路线如图2所示。首先,参照文献中的方法高效合成关键中间体——化合物1。随后,化合物1与一系列含不同取代基的苯胺衍生物发生缩合反应,以63%–79%的产率成功制得目标α-二亚胺配体L1–L5。最后,将所得配体L1–L5与(DME)NiBr2(DME = 1,2-二甲氧基乙烷)进行配位反应,以91%–96%的高产率得到相应的镍配合物Ni1–Ni5。采用核磁共振氢谱、碳谱及氟谱、元素分析、X-射线单晶衍射以及包埋体积计算等方法对所有镍配合物的结构进行了系统表征与验证。
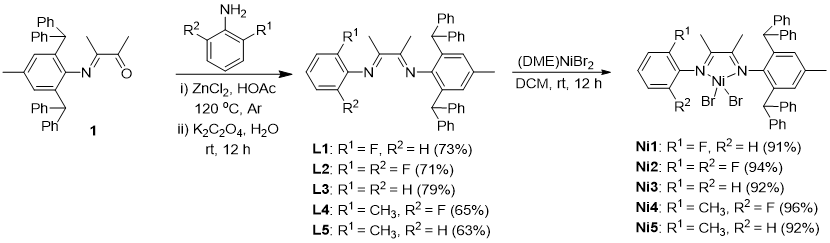
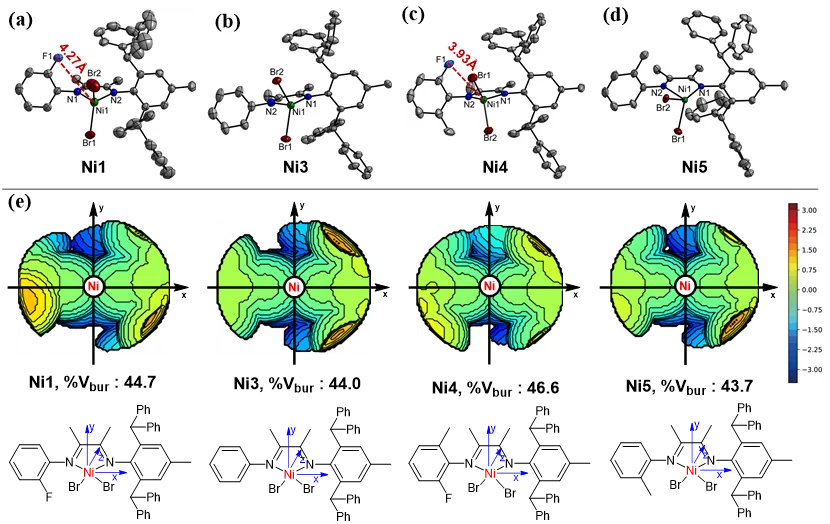
图2. α-二亚胺镍催化剂的合成
催化剂Ni1–Ni5采用非对称结构设计,其一侧保留了大位阻的二苯甲基芳胺基团,有效保证了催化剂在25–100 °C范围内的热稳定性。在Et2AlCl活化下,Ni1–Ni5在乙烯聚合中均表现出高催化活性。其中,双氟取代的Ni2在60 °C时活性最高,可达2.62 × 107 g·molNi-1·h-1。总体而言,含氟催化剂Ni1、Ni2和Ni4的聚合活性普遍高于无氟的Ni3和Ni5(图3a)。此外,双氟取代的Ni2所得到的聚乙烯分子量明显高于单氟取代的Ni1及无氟的Ni3(图3b)。推测其原因为:氟原子与聚合物增长链上的β-H之间形成的氢键作用有助于抑制β-H消除,从而延缓链转移反应。进一步引入额外的空间位阻后,Ni4可获得更高分子量的聚乙烯(Mn最高达2.99 × 105 g·mol-1),并在100 °C高温下仍保持优异的催化活性。相比之下,无氟取代的Ni5在聚合活性及所得聚合物分子量方面都稍逊于Ni4。上述结果表明,N-芳基邻位的氟效应与位阻协同策略可同时提升催化剂的聚合活性及所得聚乙烯的分子量。
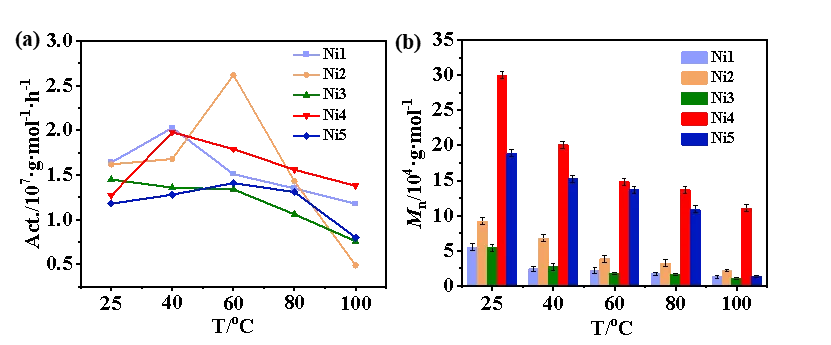
图3. Ni1-Ni5在25-100 oC下进行乙烯均聚(a)催化活性、(b)分子量的对比图
此外,本研究进一步考察了Ni1、Ni2和Ni4催化乙烯与极性单体的共聚性能。在25 °C下,上述三种催化剂均能高效催化乙烯与5-己烯-1-醇、9-癸烯-1-醇、十一烯醇及十一烯酸甲酯的共聚反应,并保持较高的聚合活性。其中,双氟取代的Ni2相较于Ni1,其极性单体插入率有所降低,但所得聚合物分子量更高。值得注意的是,兼具氟效应与位阻协同作用的Ni4表现出最优异的共聚性能,其极性单体插入率可达0.47–0.92%,聚合物分子量最高达15.31 × 104 g·mol-1,实现了插入率与分子量的同步提升。以上结果表明,在N-芳基邻位同时引入空间位阻与氟取代,不仅能有效提高共聚物分子量,还能促进极性单体的插入,为两类关键性能的协同优化提供了有效策略。
进一步地,选取Ni4与Ni5通过乙烯均聚制备的高分子量聚乙烯进行力学性能测试。结果表明,所得材料呈现典型的弹性体特征,具有优异的韧性:断裂应变可达1500–4249%,最高断裂应力达34.6 MPa,材料的弹性回复率在39–68%之间,综合性能优于文献报道的大多数聚乙烯弹性体。其中,含氟催化剂Ni4所得聚乙烯弹性体的弹性回复率高于Ni5,推测其原因为:氟效应有效抑制了β-H消除反应,促使聚合物形成更高的分子量与适宜的支化结构,从而提升弹性性能。随着聚合温度升高,材料的断裂应变显著提升,而断裂应力有所下降(图4a)。这归因于高温加速了链行走反应,导致聚合物支化度增加。更高的支化程度与分子链缠结有利于改善弹性,但同时破坏了聚合物的结晶度,从而牺牲了部分力学强度。

图4.(a)聚合物的应力-应变曲线;(b-i)聚合物样品在300%应变下10次循环的迟滞实验图
为了探究聚合物微观结构与宏观性能之间的内在关联,本研究采用高温13C NMR对Ni4在25 °C下制备的聚乙烯弹性体进行了精细结构表征。结果表明,该聚合物以甲基支链为主(甲基支链占比71.8%),同时含有18.8%的长链支链,充分体现了催化剂具备强效的“链行走”能力。长支链的存在有利于形成有效的分子链缠结网络,从而赋予材料优异的弹性性能(弹性回复率SR = 58%)。
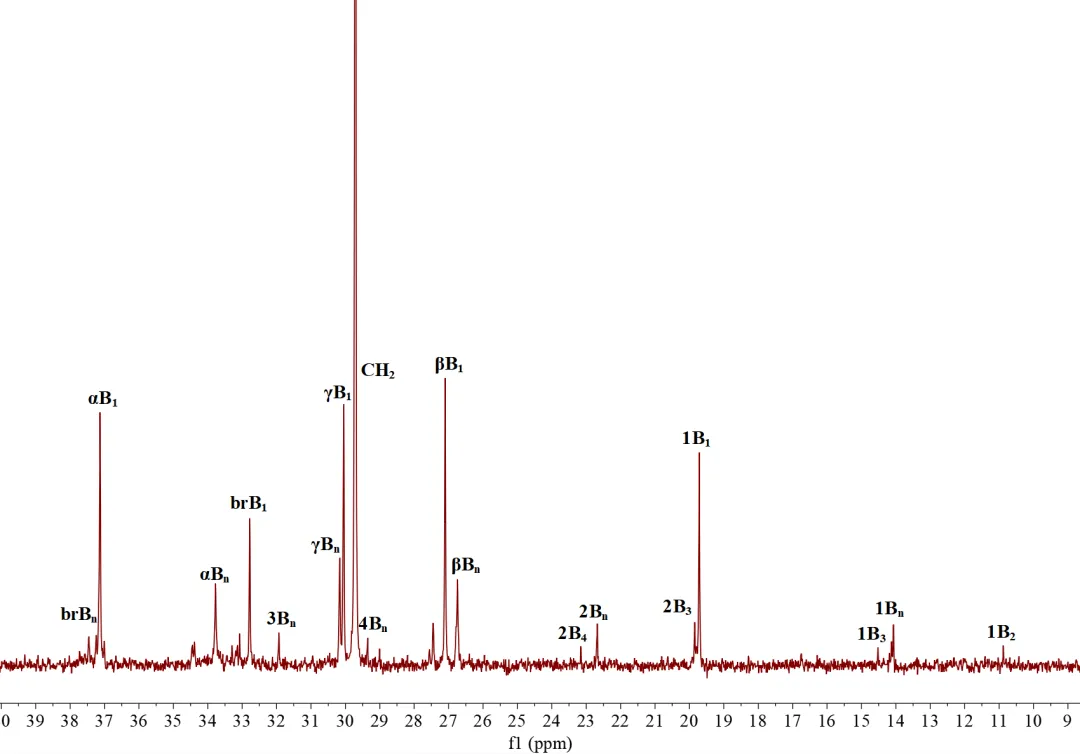
图5. 25oC下Ni4制备的聚乙烯弹性体的13C NMR
综上所述,面向功能化聚烯烃的合成需求,本研究成功开发了一类基于大体积二苯甲基骨架与N-芳基邻位氟取代相结合的非对称α-二亚胺镍催化剂。其中,氟取代催化剂Ni1、Ni2和Ni4表现出优异的乙烯均聚活性与共聚性能。研究进一步揭示了位阻效应与氟效应的协同作用机制,阐明了二者对催化性能、聚合物微观结构及材料宏观性能的关键调控规律。
该项工作得到了国家自然科学基金面上项目(No. 22471247),国家自然科学基金联合(重点)基金(No. U1904212),河南省自然科学基金(No. 222300420294)等项目资助。
Front Cover forPolymerScience & Technology:

设计灵感:在这幅以浩瀚宇宙为背景的封面图中,自主研发的镍系催化剂被置于视觉核心。该催化剂作为分子转化的关键引擎,驱动着多样化乙烯基弹性体与极性聚烯烃材料的高效合成。这些高端聚烯烃材料构成了现代工业的基石,广泛应用于汽车制造、光伏封装、5G/6G通信及光纤系统等前沿领域。
论文信息:
Unsymmetrical Fluorinated α-Diimine Nickel Catalysts with Synergistic Steric and Fluorine Effects: toward Ethylene-Based Elastomers and Polar Copolymers
Jin-Kui Liu, YanruFeng, JiayuNiu, Huizhu Wang, Mao-Ping Song, Jun-Fang Gong, * and Hui Jiang*
Polym. Sci. Technol.2026,
https://doi.org/10.1021/polymscitech.5c00139.
相关进展
郑州大学姜辉/龚军芳、中科大邹陈 CEJ:为聚烯烃赋能 - 兼具荧光、光响应、自修复与信息加密功能的智能材料
中国科大陈昶乐/邹陈团队 Angew:多重刺激响应聚烯烃基驱动器

高分子科技原创文章。欢迎个人转发和分享,刊物或媒体如需转载,请联系邮箱:info@polymer.cn


诚邀投稿
欢迎专家学者提供稿件(论文、项目介绍、新技术、学术交流、单位新闻、参会信息、招聘招生等)至info@polymer.cn,并请注明详细联系信息。高分子科技®会及时推送,并同时发布在中国聚合物网上。
欢迎加入微信群 为满足高分子产学研各界同仁的要求,陆续开通了包括高分子专家学者群在内的几十个专项交流群,也包括高分子产业技术、企业家、博士、研究生、媒体期刊会展协会等群,全覆盖高分子产业或领域。目前汇聚了国内外高校科研院所及企业研发中心的上万名顶尖的专家学者、技术人员及企业家。
申请入群,请先加审核微信号PolymerChina(或长按下方二维码),并请一定注明:高分子+姓名+单位+职称(或学位)+领域(或行业),否则不予受理,资格经过审核后入相关专业群。

点
这里“阅读原文”,查看更多

