郑州大学生命科学学院丨SLC25A3 缺陷致肥厚型心肌病:线粒体移植可逆转钙稳态失衡与心肌肥厚
- 2026-05-28 14:52:43
文献学习
本研究以 SLC25A3 敲除及错义突变(c.C544T、c.A547G、c.C349T)人诱导多能干细胞心肌细胞为模型,首先证实 SLC25A3 缺失 / 突变可重现心肌肥厚、舒张功能下降、钙稳态失衡表型;进而发现其通过损伤线粒体形态与氧化磷酸化、降低 ATP 合成、激活 AMPK 并代偿性增强糖酵解,导致乳酸与 H⁺堆积,激活 NHE1–NCX1 通路引发钙超载,进而激活 CAMKII–HDAC 与 NFAT 通路促肥厚;最后证明线粒体移植可提升氧化磷酸化、抑制过度糖酵解、恢复钙稳态并改善心肌肥厚,为 SLC25A3 相关肥厚型心肌病提供机制解释与潜在治疗策略。

研究背景
肥厚型心肌病是心衰与心源性猝死常见病因,除肌节基因外,线粒体相关基因突变可致线粒体性肥厚型心肌病;SLC25A3 编码线粒体磷酸载体 PiC,其纯合或复合杂合突变临床可引发乳酸酸中毒、心肌肥厚与早亡,但致病机制不清,且人与小鼠心脏生理代谢存在差异,亟需人类心肌模型阐明机制并探索疗法。
研究方案
利用 CRISPR‑Cas9 构建 SLC25A3 敲除及 c.C544T、c.A547G、c.C349T 错义突变 hiPSC‑CM 模型,先验证是否重现疾病表型,再解析线粒体能量代谢、钙稳态、糖酵解及下游信号通路的改变,明确关键致病链,最后通过线粒体移植干预,验证其对表型与分子异常的挽救效果,同时评估突变致病性并指出研究局限。
研究结果
SLC25A3 敲除心肌细胞呈现心肌肥大、肌节紊乱、肥厚基因上调、收缩舒张异常与钙稳态失衡;同时出现线粒体碎裂、嵴破坏、膜电位下降、ROS 升高、ATP 合成降低、复合物 IV/V 活性下降,糖酵解显著增强并堆积乳酸与 H⁺,激活 NHE1‑NCX1 引发钙超载并活化 CAMKII‑HDAC/NFAT 通路;三种错义突变可诱发与敲除相似表型,而正常 SLC25A3 过表达无异常;线粒体移植可有效恢复线粒体功能、逆转糖酵解亢进、纠正钙稳态并改善心肌肥厚。
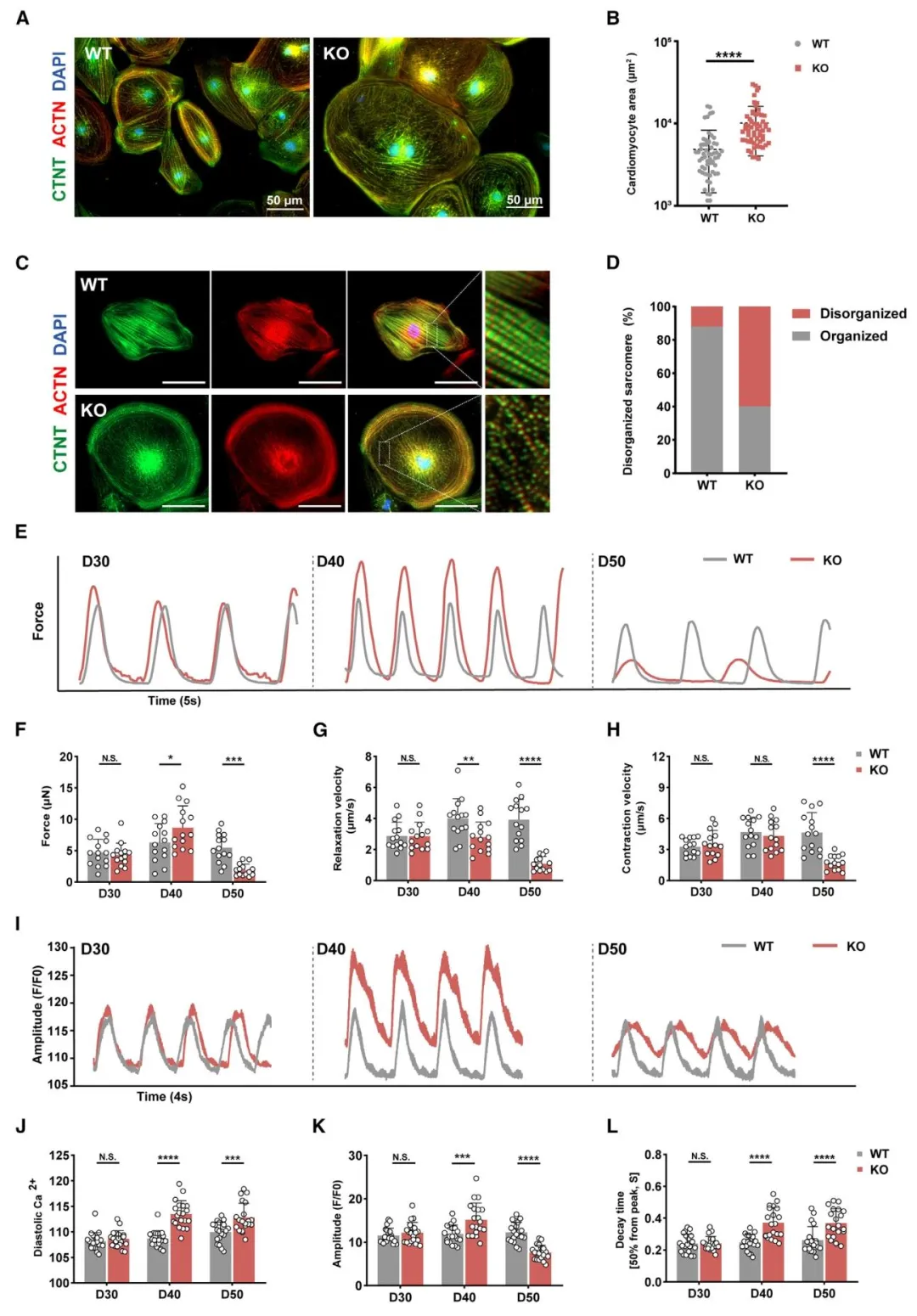
Figure1:证实 SLC25A3 敲除 hiPSC‑CM 成功模拟心肌肥厚、收缩舒张功能障碍及钙稳态失衡表型。
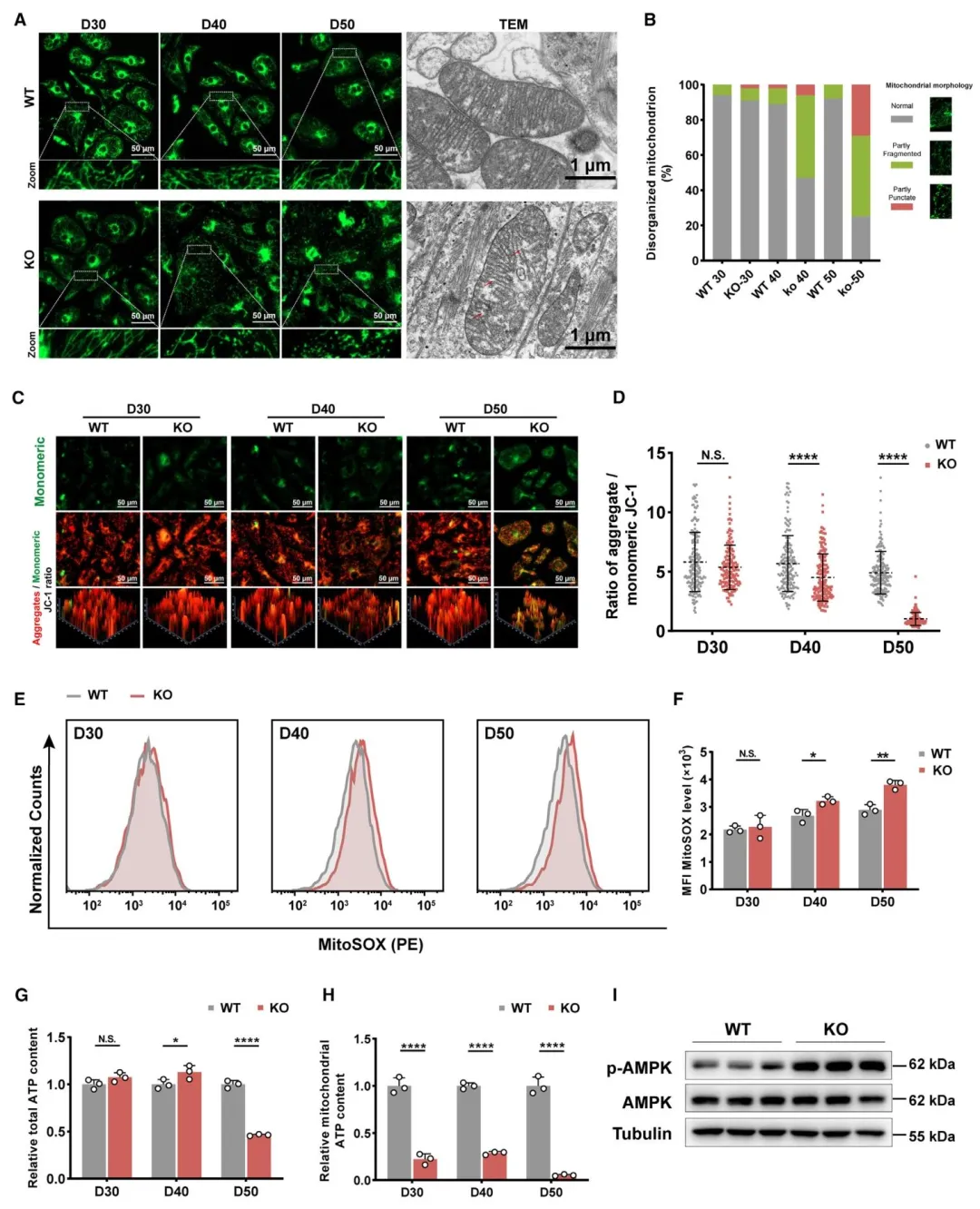
Figure2:揭示 SLC25A3 缺失导致线粒体形态破坏、功能损伤、ATP 合成下降及能量应激激活。
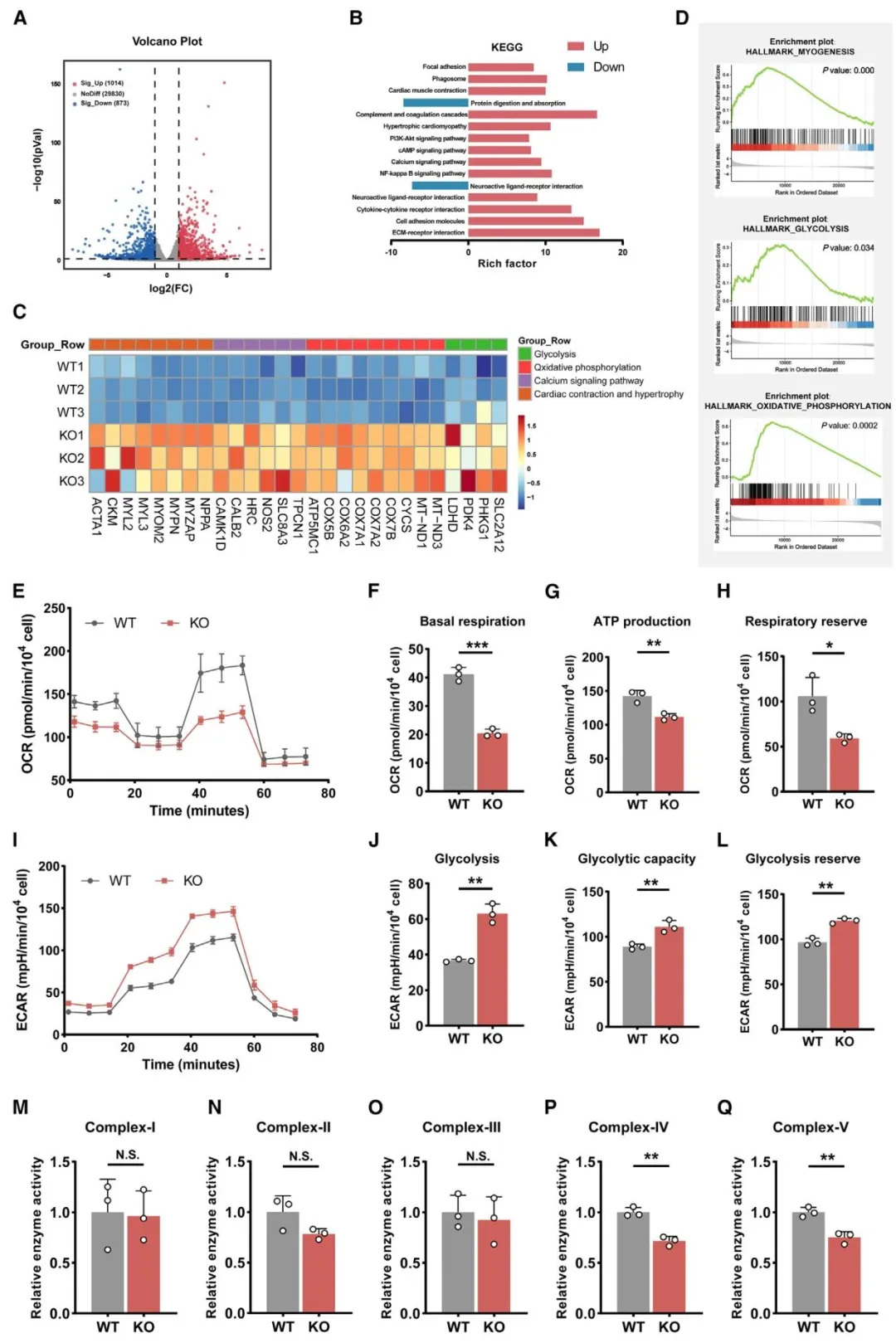
Figure3:表明 SLC25A3 缺陷上调钙信号与肥厚通路、抑制氧化磷酸化、显著增强糖酵解。
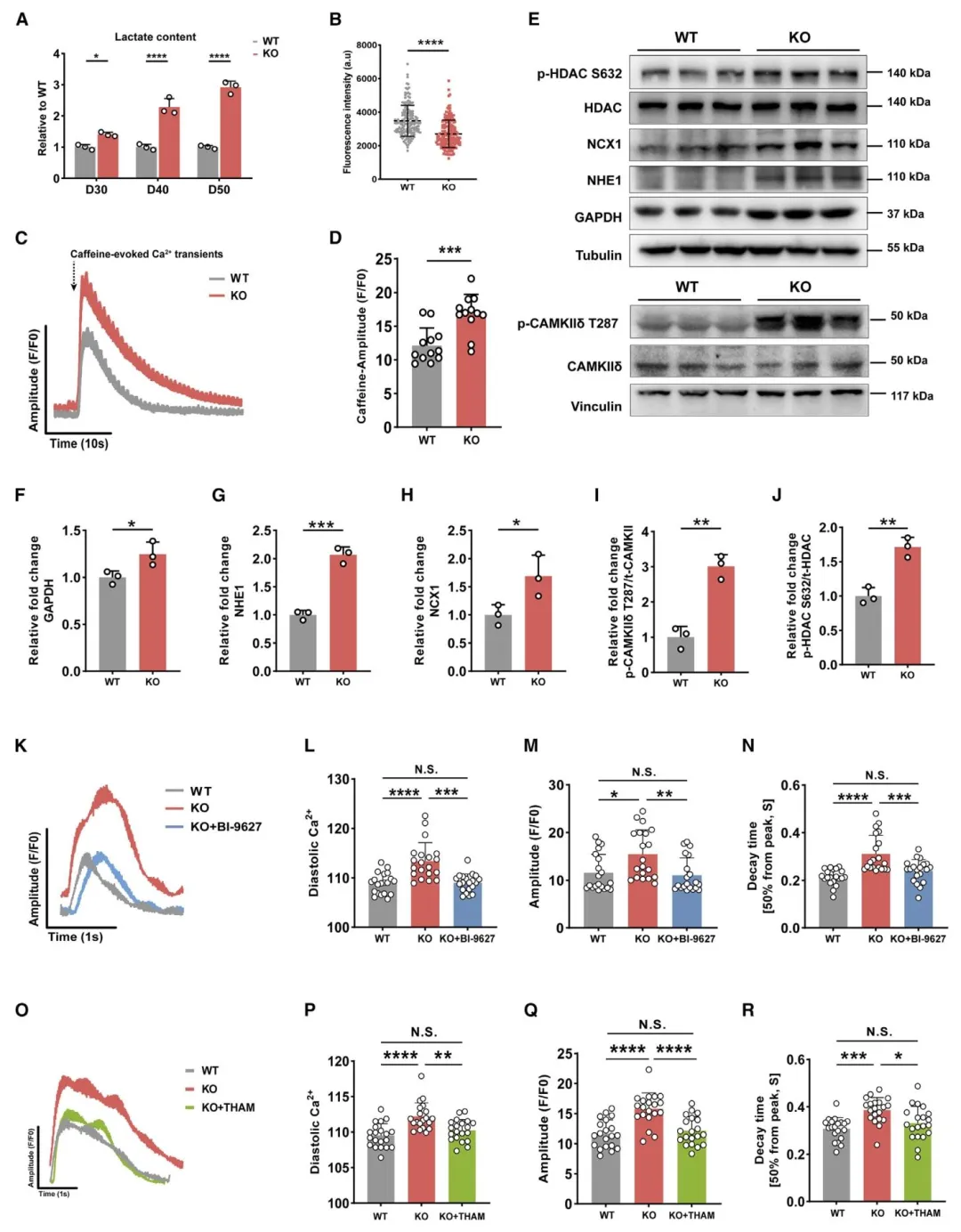
Figure4:证明糖酵解副产物 H⁺堆积通过 NHE1‑NCX1 介导钙稳态失衡,是肥厚重要机制。
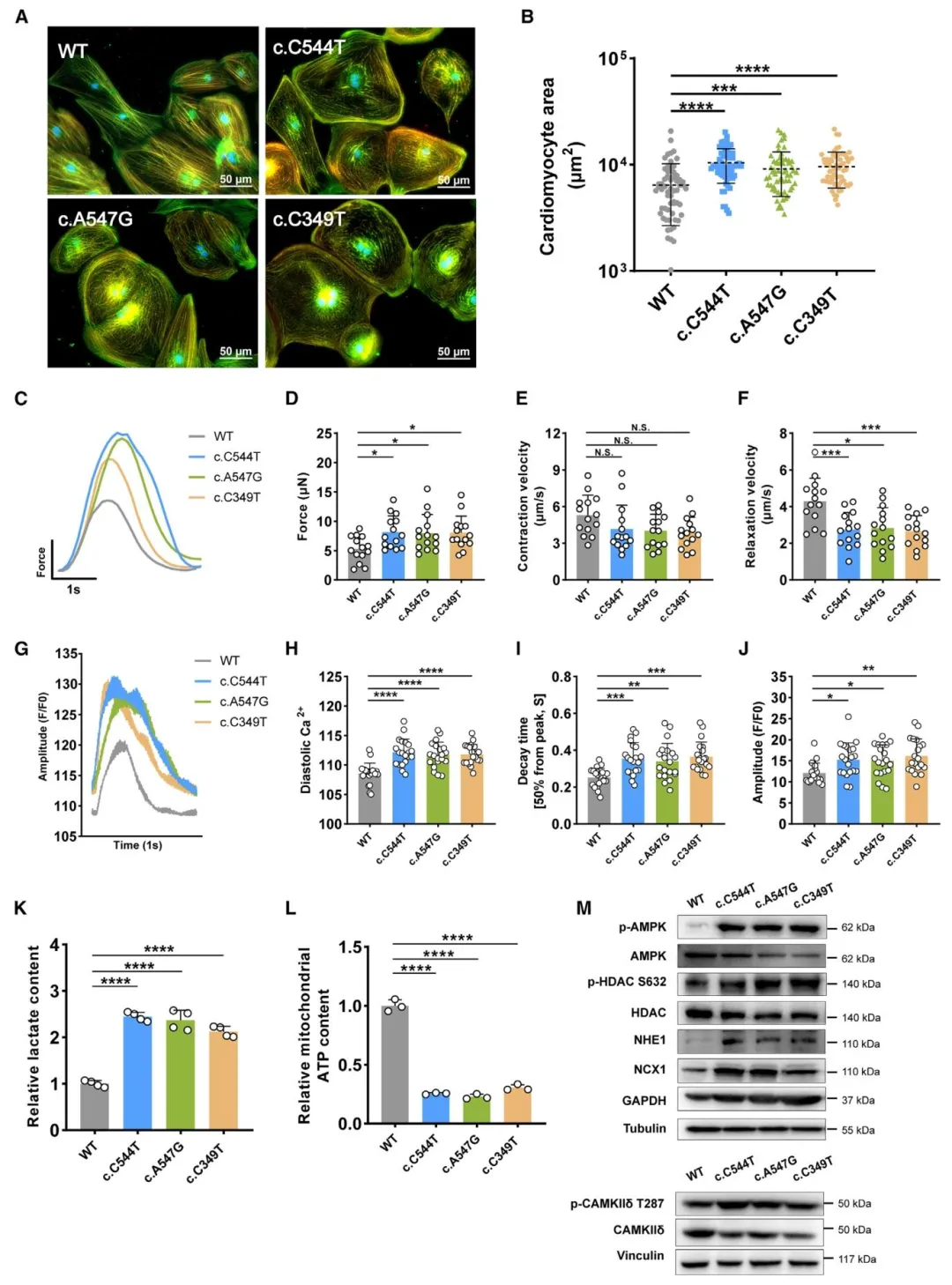
Figure5:验证 c.C544T、c.A547G、c.C349T 错义突变可诱发与敲除一致的疾病表型。
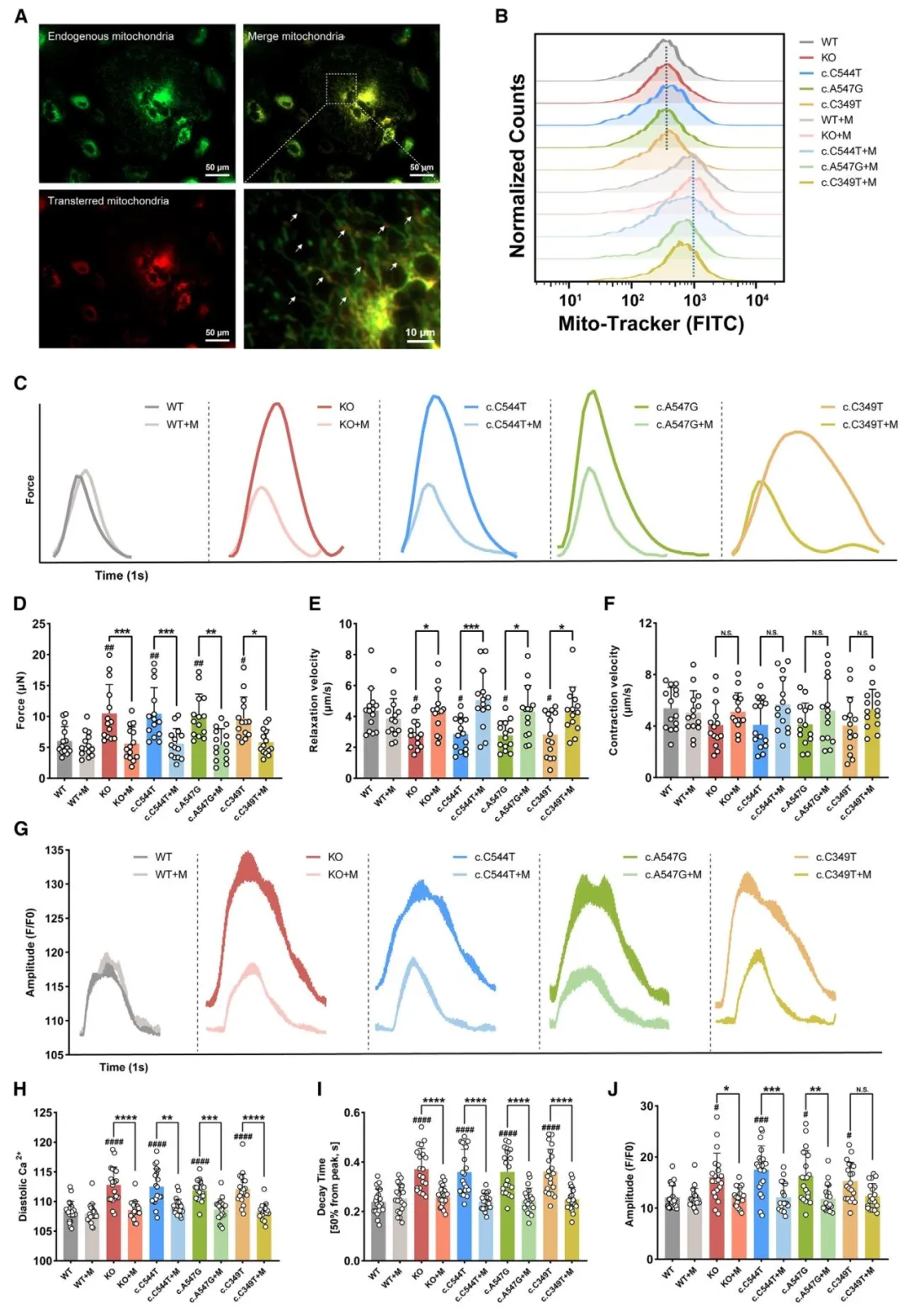
Figure6:显示线粒体移植可成功进入细胞并挽救心肌收缩功能与钙瞬变异常。
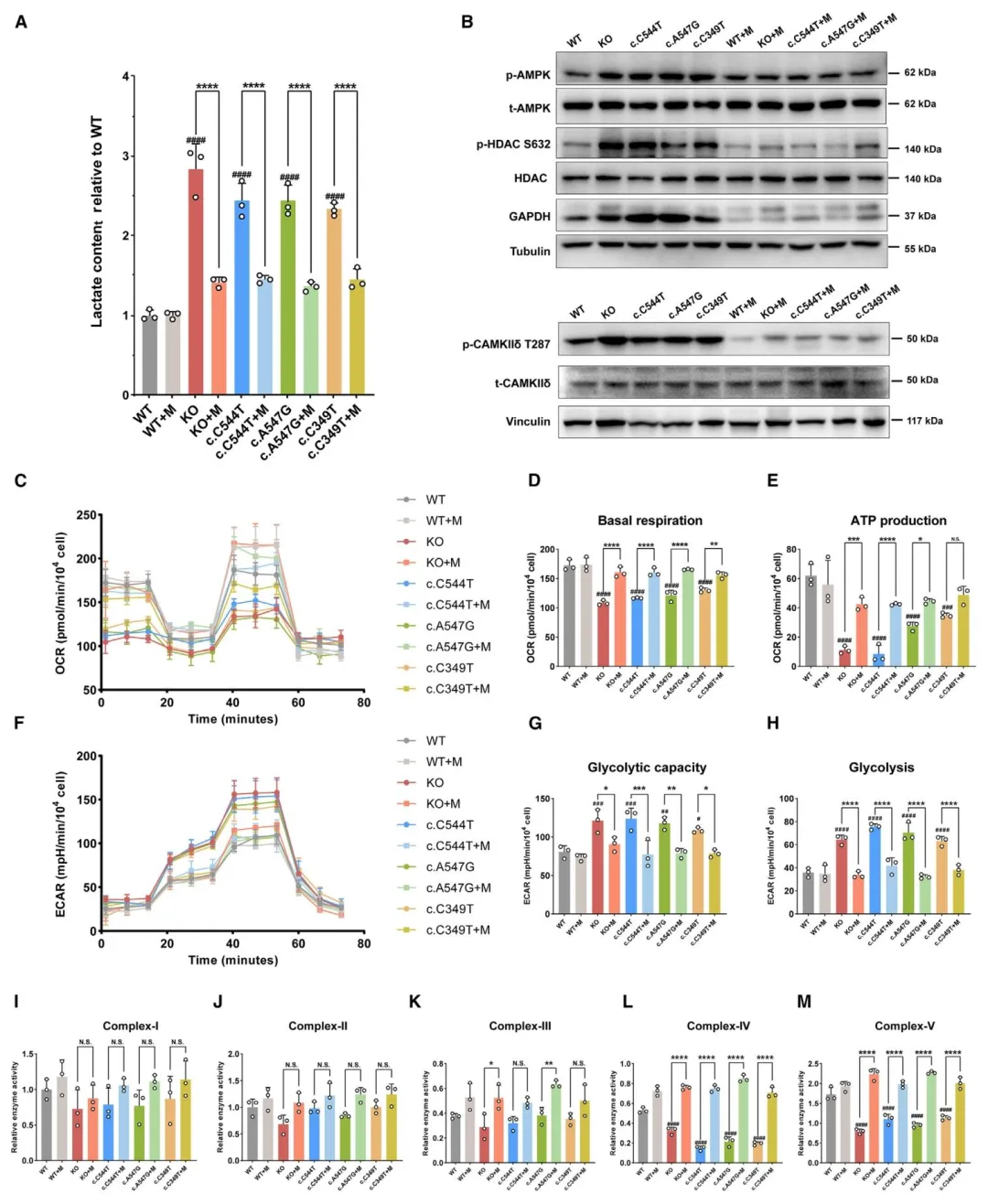
Figure7:阐明线粒体移植通过改善线粒体呼吸、抑制糖酵解、阻断钙信号通路发挥疗效。
研究结论
本研究阐明 SLC25A3 缺陷 / 错义突变通过线粒体能量不足→糖酵解代偿亢进→H⁺堆积→NHE1‑NCX1 激活→钙稳态失衡→肥厚信号通路活化的链式机制诱发肥厚型心肌病,明确 c.C544T 等三种突变具致病性,并证实线粒体移植可有效逆转异常;局限性在于未获取患者原代细胞、未在器官水平验证、未完全解析乳酸的具体作用,hiPSC‑CM 模型也无法完全模拟体内复杂环境。
本文中使用的图片来源Pubmed,因客观原因未能与权利人取得联系。本平台出于学术交流目的引用,无意侵犯原作者权益。如权利人认为不妥,请及时联系公众号后台,我们将立即删除或协商解决。
医学国自然,省自然,博士课题设计,医学实验外包,医学SCI,实验方案设计,免费的线上博导一对一沟通,确认实力后再谈合作,科研合作可以加微信:SCI971SCI

国家杰青一对一答疑视频
医学省自然申请答疑,立项的关键条件是哪一些?从哪些方向可以杀出重围
临床型博士如何准备国青标书?没有预实验怎么办?专家一对一解答规划
中医药科研研究