
导读
绿色氢能是实现“双碳”目标的关键纽带。然而,作为阴离子交换膜电解水(AEMWE)的核心反应,阳极析氧反应(OER)极为迟缓。传统催化剂在工业级大电流密度下,往往面临“活性与稳定性不可兼得”的残酷博弈。近日,《Angewandte Chemie International Edition》在线发表了来自郑州大学的最新研究成果。科研团队通过在超薄氧化铁纳米片中巧妙进行原子级碳掺杂,成功打破了这一历史性难题,实现了兼具超高活性与超长寿命的工业级电解水催化剂,这为规模化绿色制氢扫清了关键障碍。
一、研究背景:工业级绿氢制备的“阿喀琉斯之踵”
在应对全球化石能源枯竭与环境污染的战役中,利用可再生能源电解水制取绿氢已被视为未来清洁能源技术的核心演进方向 。其中,阴离子交换膜电解水(AEMWE)技术由于可以使用非贵金属组分作为电极材料,在降低设备制造成本和提高能量转换效率方面展现出了巨大的竞争潜力 。
然而,理想很丰满,现实很骨感。作为电解水过程中的“发动机”,阳极析氧反应(OER)由于涉及复杂的四电子转移过程,其反应动力学极其迟缓,极大地拉低了整体的能量效率 。虽然传统的贵金属催化剂(如二氧化钌和二氧化铱)在高电流密度下具有不错的活性,但其资源稀缺、价格高昂,根本无法满足未来工业化规模的应用需求 。
因此,科学家们将目光转向了储量丰富的过渡金属氧化物(如氧化铁)。遗憾的是,目前的改性策略主要围绕两种机制展开,但它们都走入了死胡同:
1.吸附演化机制(AEM): 依赖优化 *OH、*O、*OOH 等中间体的吸附能 。但在该机制下,*OH 和 *OOH 的结合能之间存在难以打破的“线性标度关系”,导致理论析氧过电位被死死限制在 370 mV 左右,活性很难获得质的突破 。
2.晶格氧介导机制(LOM): 引入晶格氧参与反应以大幅提升本征活性 。但频繁的晶格氧参与会导致催化剂表面产生大量的氧空位,引发本体氧向外动态迁移,最终导致催化剂结构崩溃和金属位点严重溶解 。
为了维持催化剂的稳定性,目前普遍的做法是抑制晶格氧(LOM)路径,但这又是以牺牲活性为代价的 。这种“活性提高、稳定性下降”或“稳定性提升、活性骤减”的矛盾博弈,成为了阻碍非贵金属催化剂迈向工业化应用的“阿喀琉斯之踵” 。 为了彻底打破这一魔咒,郑州大学研究团队另辟蹊径,将目光聚焦于激活氧化物路径机制(OPM) 。理论上,OPM 机制能够通过高密度的 *OH 覆盖度,促使 *O 自由基直接进行偶联产生氧气,完全不需要晶格氧的牺牲,也无需经历 *OOH 中间体,能完美兼顾高活性与高稳定性 。然而,在实验上如何在非贵金属表面精准构筑并触发 OPM 路径,一直是国际催化界的一大挑战 。
二、研究内容:小碳原子激发出惊人的工业级催化本领
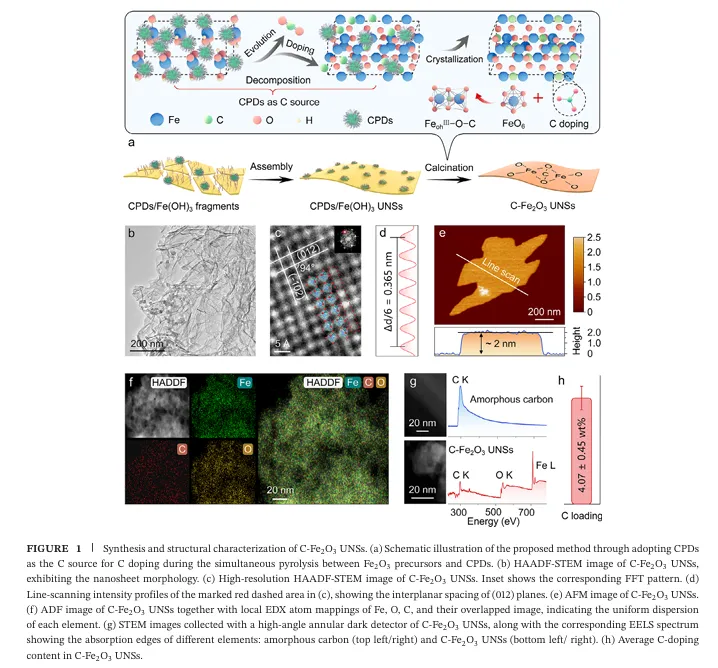
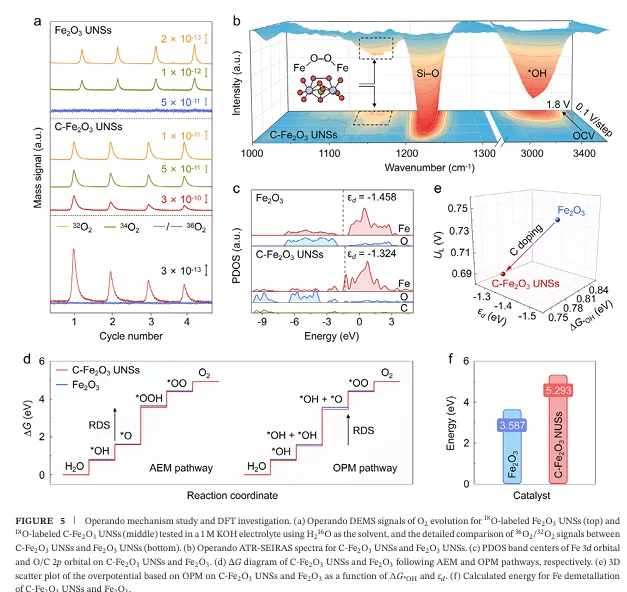
在这项工作中,研究团队开发出了一种利用碳化聚合物点(CPDs)作为碳源的界面局域外延生长及原位热解策略 。在 500 oC 的焙烧过程中,超薄纳米片表面的 CPDs 分解,促使原子级的碳(C)精准掺杂进入 Fe2O3 超薄纳米片的八面体空隙中,成功制备了具有原子级碳掺杂的氧化铁超薄纳米片(简称为 C- Fe2O3 UNSs) 。该纳米片的厚度仅为约 2.0 nm 。
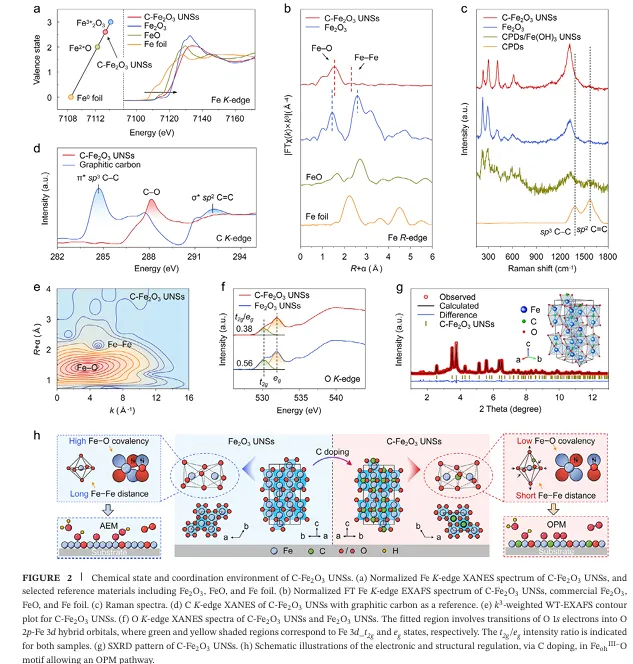
多项先进表征(如同步辐射 X 射线吸收谱 XANES/EXAFS)表明,掺杂的 C 原子在氧化铁晶格中并非以石墨碳或无定形碳的形式存在,而是形成了独特的 FeohIII-O-C 桥连配位构型,显著调控了邻近 FeohIII中心位点的电子结构 。正是这种精准的微观结构调控,为其带来了极度震撼的催化反应活性数据:
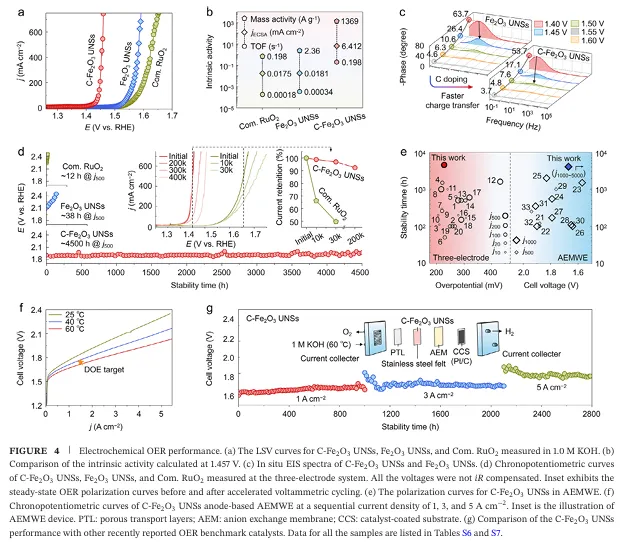
·超低的过电位与卓越的本征活性: 在 1.0 M KOH 碱性电解液中,C- Fe2O3 UNSs显示出极其优异的 OER 活性 。它仅需要 227 mV 的超低过电位,即可驱动高达 500 mA cm -2的工业级大电流密度j500 ! 这一数值不仅比未掺杂的纯 Fe2O3 纳米片(354 mV)降低了 36% ,更是远远优于商业贵金属催化剂 RuO2(402 mV)和传统的泡沫镍(708 mV) 。此外,该催化剂在 j500 下的法拉第效率接近 100%,证明高电流完全来自于析氧反应本身 。
·无惧严苛考验的惊人耐力(4500小时稳定性): 在模拟工业级高极化电流密度的恒电位(CP)测试中,C- Fe2O3 UNSs展示出了非凡的稳定性 。在 500 mA cm-2 的超大电流密度下连续运行超过 4500 小时(约6个月),电压仅微幅上升了 68 mV,对应的电压降解速率仅为极低的 15 mV kh-1 ! 相比之下,传统的纯 Fe2O3 和商业 RuO2 分别在运行短短 38 小时和 12 小时后就彻底失活崩溃 。此外,催化剂在经历了高达 20 万、30 万甚至 40 万次连续循环伏安(CV)扫描后,极化曲线仍近乎重合,表现出铁壁一般的结构耐受力 。
·全电解水器件中的标杆级表现(AEMWE测试): 研究人员进一步将该催化剂作为阳极组装进阴离子交换膜电解水(AEMWE)实用器件中 。在不进行 iR 补偿的情况下,器件仅需 1.72 V 的电池电压即可驱动 1.5 A cm-2 的超高全电池电流密度,轻松跨过了美国能源部(DOE)的 2025 目标线 ! * 工业阶梯式老化测试(2800小时): 在 1 至 5 A cm-2 的极端大范围电流密度阶梯式加速老化测试中,该器件稳定运行了 2800 小时 。特别是在 3 A cm-2 的超大工业电流密度下,其全电池电压几乎保持恒定,估计电压以降解率仅为 3.05 mV kh-1 的极低速率衰减 ,这完全达到了美国能源部(DOE)对于未来实用化绿氢制备的苛刻指标 。
三、工作创新点:精准调控电子结构,开辟催化新路径
本项研究的核心魅力不仅在于刷新了多项性能纪录,更在于它从底层物理化学机制上给出了科学的创新解答:
1.首次在非贵金属氧化物中实现原子级碳掺杂与稳定: 传统碳掺杂极易形成非晶碳包裹或碳聚集体 。该工作巧妙地利用 CPDs 作为碳源,使 C 原子能够精准进入 FeO6 八面体空隙中,通过构筑稳定的 FeohIII-O-C 配位形态进行“安家” 。这种策略不仅降低了 Fe 的平均价态(介于 +2到 +3 之间) ,还上移了 Fe 的 3d 轨道,实现了金属中心局域电子结构的精细化定制 。
2.巧妙降低 Fe-O 共价性,完美改写 OER 反应路径: 原位测试与理论计算证实,FeohIII-O-C桥连键的形成削弱了 O 2p 与 Fe 3d 轨道的杂化,降低了 Fe-O 的共价性 。这一关键变化带来了双重红利:一方面,它在物理上极大地抑制了晶格氧的溶解和外迁,从根本上保证了催化剂的长期结构稳定 ;另一方面,它构建出了高效的 FeohIII-O- FeohIII 双铁协同活性中心,该中心能够极大富集界面 *OH 的吸附覆盖度,将反应路径从传统的 AEM 机制强行切换为 OPM( Oxide Pathway Mechanism,氧化物路径机制) 。
3.开创直接 *O-*O 自由基偶联,打破线性标度限制: 借助同位素标记的产物差分电化学质谱以及原位衰减全反射表面增强红外吸收光谱(Operando ATR-SEIRAS),团队清晰抓取到了反应过程中产生的特征桥连氧中间体(*O-*O) 。这意味着相邻位点吸附的 *O 自由基可以直接发生能量壁垒极低的“偶联”并释放出氧气,绕过了产生 *OOH 的高能垒步骤 。正是反应路径的彻底改变,使其彻底摆脱了线性标度关系的束缚,打破了“活性-稳定性”互为牺牲的僵局 。
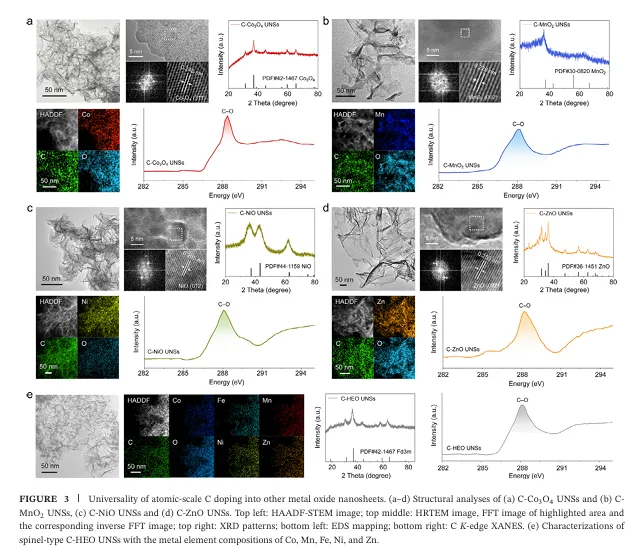
4.方法具备广阔的“普适性扩展空间”: 更令人振奋的是,这种原子级碳掺杂策略具有极高的普适性 。团队证明该方法不仅能应用在氧化铁上,还能同样成功地在超薄纳米片结构的 Co3O4、MnO2、NiO、ZnO 乃至尖晶石型高熵过渡金属氧化物(HEOs)以及钙钛矿等多种催化剂中实现均匀的原子级碳掺杂 。这为未来跨体系设计高性能、多功能的长寿命工业级电催化剂开辟了一扇全新的大门 。
原文信息
Z. Hu, J. Yu, and J. Chang, et al.,Ultrastable Non-Noble-Metal Oxygen Evolution Electrocatalyst for Industrial-Level Water Electrolysis, Angewandte Chemie International Edition (2026): e2311940, https://doi.org/10.1002/anie.2311940.
声明:本文内容用于科研进展分享,详细内容请参考原文,如有侵权,请联系后台小编删除。
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
