郑州大学 Adv. Sci. | 三核铜簇碳硼烷COF实现高能快速自燃点火
商业合作 / 安瓿瓶采购 / 加入 COFs & MOFs 交流群,欢迎添加小编微信:COFsandH2O2
郑州大学 Adv. Sci. | 三核铜簇碳硼烷COF实现高能快速自燃点火
自燃材料可在接触氧化剂后自发点火,是航天推进和国防系统中的重要燃料类型。传统肼类燃料虽具有较高比冲,但存在毒性高、体积能量密度低和操作风险大等问题。硼富集化合物具有较高能量密度,其中碳硼烷因三维笼状结构和热稳定性受到关注,但其化学惰性限制了直接自燃点火。针对“高能量单元难以快速点火”和“金属簇催化位点易被配体屏蔽”的问题,作者将三核铜簇作为结构节点和催化中心,同时引入醛基功能化碳硼烷作为高能连接单元,构筑了铜簇基共价有机框架Cu3-CB-COF,实现催化中心与能量单元在同一框架中的协同整合。
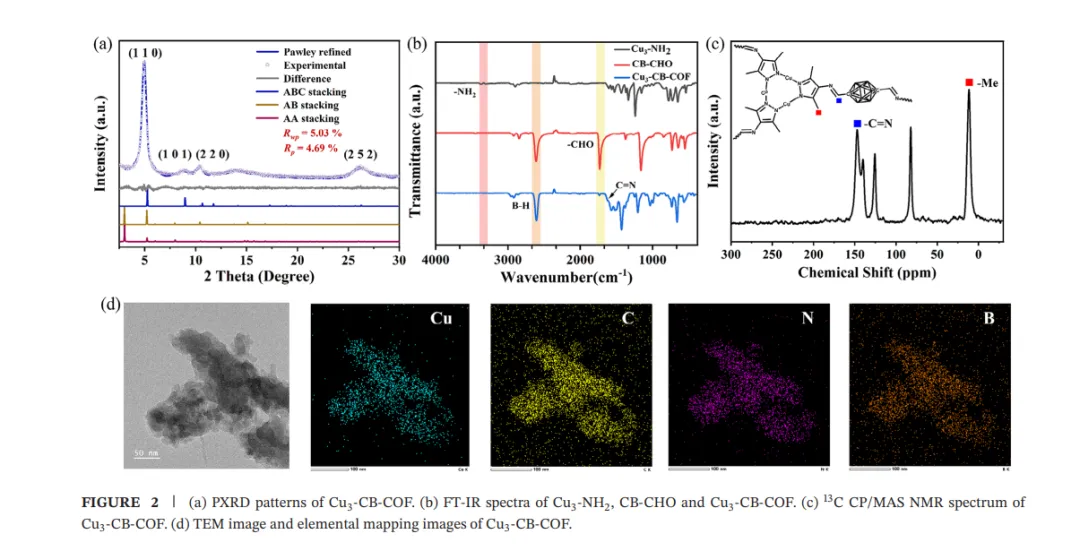
Cu3-CB-COF由Cu3-NH2与CB-CHO通过Schiff碱缩合反应合成。PXRD显示Cu3-CB-COF在2θ = 5.45°处出现对应(110)晶面的强峰,并在9.19°、10.71°和26.13°处出现其他衍射峰;Pawley精修支持ABC堆积模型,空间群为R3̅,晶胞参数为a = b = 32.7679 Å、c = 10.3581 Å,Rp = 4.69%,Rwp = 5.03%。FT-IR中N–H和–CHO伸缩振动消失并出现C=N伸缩振动,说明亚胺键形成;B–H伸缩振动保持不变,表明碳硼烷笼结构在框架中保持完整。固体13C CP/MAS NMR在148 ppm处显示C=N信号,XPS中B 1s和N 1s信号进一步支持碳硼烷和氮配位环境的存在。XAFS结果显示Cu3-CB-COF中Cu价态接近+1,且相较Cu3-NH2未发生明显吸收边位移,说明COF组装未明显改变三核铜中心的价态与结构;R空间约1.5 Å处峰对应Cu–N配位。N2吸附–脱附结果显示Cu3-CB-COF的BET比表面积为421.1 m² g−1,孔径分布包含0.51和1.88 nm微孔以及3.86 nm介孔;TEM和元素映射显示Cu、C、N和B在材料中均匀分布。

以高浓度过氧化氢HTP为氧化剂进行标准氧化剂–燃料液滴测试时,未组装的Cu3-NH2点火延迟时间为60 ms,但燃烧仅产生分散且较弱的火花;Cu3-CB-COF的点火延迟时间缩短至40 ms,并产生约7 cm高的明亮红绿色火焰。将10 mg Cu3-CB-COF填入石蜡蜡粒中心后,与HTP接触70 ms发生自燃反应,并伴随石蜡熔化和燃烧,说明其可作为复合推进剂中的反应性添加剂。能量参数方面,Cu3-CB-COF的标准摩尔燃烧焓为−119323.6 kJ mol−1,质量能量密度为25.9 kJ g−1,在最优氧燃比O/F = 4时最大比冲为250.5 s;相比之下,Cu3-NH2的标准摩尔燃烧焓为−7669.5 kJ mol−1,质量能量密度为14.7 kJ g−1,最大比冲为235.5 s。对照材料Cu3-Ph-COF将CB-CHO替换为对苯二甲醛后,点火延迟时间延长至67 ms,质量能量密度降至19.9 kJ g−1,说明碳硼烷单元对提升能量输出具有直接贡献。PXRD结果还显示,Cu3-CB-COF、Cu3-Ph-COF和Cu3-NH2在水中放置14 d以及空气中放置30 d后均保持原有晶相。
机理方面,作者通过理论计算分析了Cu3-NH2和Cu3-CB-COF与H2O2之间的界面相互作用。差分电荷密度显示H2O2吸附在Cu位点后发生由金属中心向氧化剂的电子转移;Bader电荷分析表明,吸附在Cu3-CB-COF上的H2O2获得0.1170 e−净电荷,高于Cu3-NH2体系,说明碳硼烷框架不仅保留三核铜中心的电子转移能力,还增强了界面电子转移。在反应路径计算中,H2O2经初始吸附后在金属核心发生O–O键断裂并形成羟基自由基,Cu3-CB-COF对应的过渡态能垒低于Cu3-NH2,最终态也更稳定,表明其反应动力学和中间体稳定性更有利。扩散能垒计算进一步显示,H2O2靠近Cu3-NH2和Cu3-CB-COF吸附位点时未出现明显扩散能垒,而大核数铜簇因三维拥挤配体壳层产生空间位阻,限制氧化剂接近金属中心。该研究表明,将暴露的三核铜催化节点与碳硼烷高能连接单元整合进COF骨架,可同时实现快速氧化剂活化和高能量释放,为固体自燃燃料的框架化设计提供了新的思路。
https://doi.org/10.1002/advs.76306
本文使用科应智能助手(www.scienceing.com)开展文献调研


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
