郑州大学,第一单位,校史首篇Nature Catalysis!
- 2026-07-04 22:23:29


第一作者:Chao Lei, Wen Lu, Tingting Shen
通讯作者:陈学年、马艳娜、魏东辉
通讯单位:郑州大学
1、研究背景:碳硼烷(C₂B₁₀H₁₂)作为具有三维芳香性的硼簇化合物,在材料化学、药物研发及硼中子俘获治疗领域展现巨大潜力。碳硼烷含硼量高,是硼中子俘获治疗的潜在候选材料。开发碳硼烷引入有机分子的新方法具有重要意义。
2、核心难点:由于B-H键极性弱且存在十个等效B-H键,其硼顶点选择性官能化尤其是不对称催化反应仍极具挑战。传统迈克尔加成反应可高效构建β-手性中心,但α-手性中心的逆向反迈克尔加成反应却鲜有报道。
3、研究切入点:基于前期发现——软亲电体Pd(II)可优先活化碳硼烷B-H键,提出假设:通过Pd(II)与手性磷酸(CPA)形成手性催化剂,可能实现碳硼烷作为亲核试剂与α,β-不饱和羧酸的逆向加成。选择HFIP为溶剂可促进B-H键活化,并以间位碳硼烷为模型底物进行条件筛选。
1.规律推测:在钯(II)催化条件下,碳硼烷与α,β-不饱和羰基化合物有望发生不对称迈克尔加成反应。
2.共同催化:在乙酸钯与手性磷酸(CPA)的共同催化下,碳硼烷与α,β-不饱和羧酸反应可高选择性生成α-加成产物。
3.机理验证:结合实验与计算化学研究,阐明了该反应的机理及对映选择性的来源,证实稳定五元钯环中间体的形成是实现逆向选择性的关键。
图 1| 碳硼烷的应用以及催化不对称迈克尔加成和反迈克尔加成
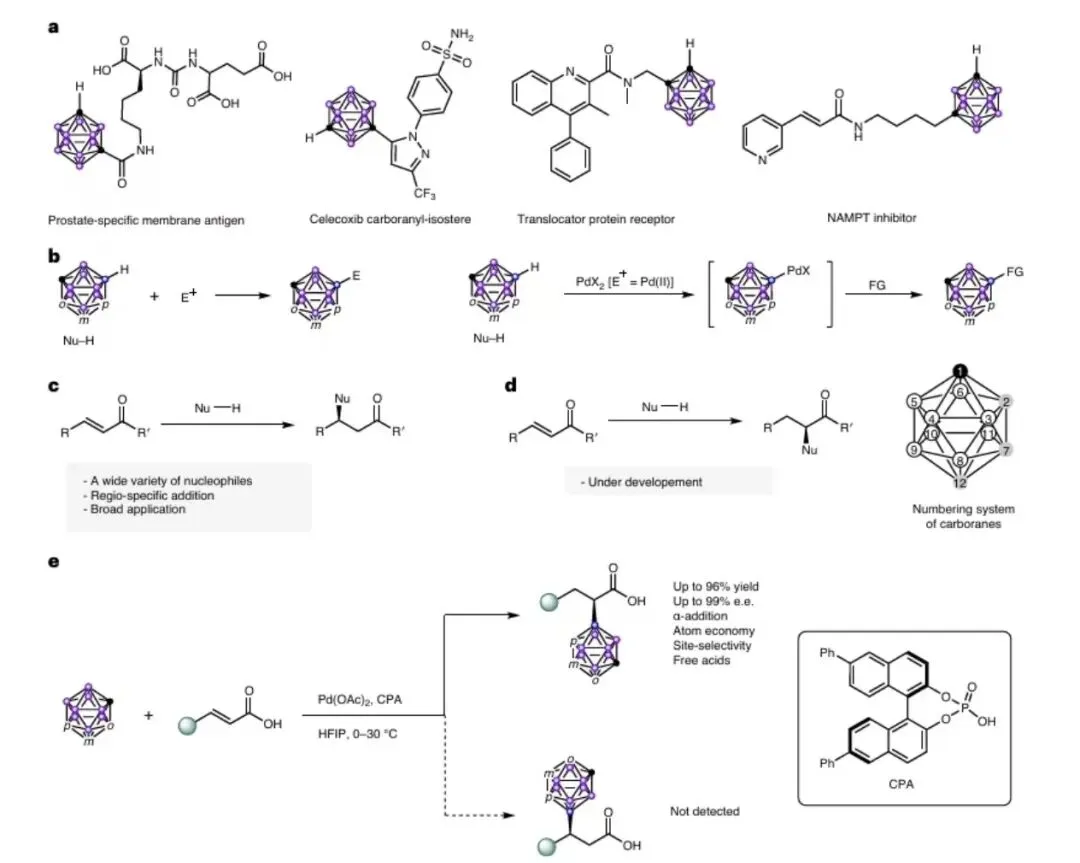
图 2|配体筛选
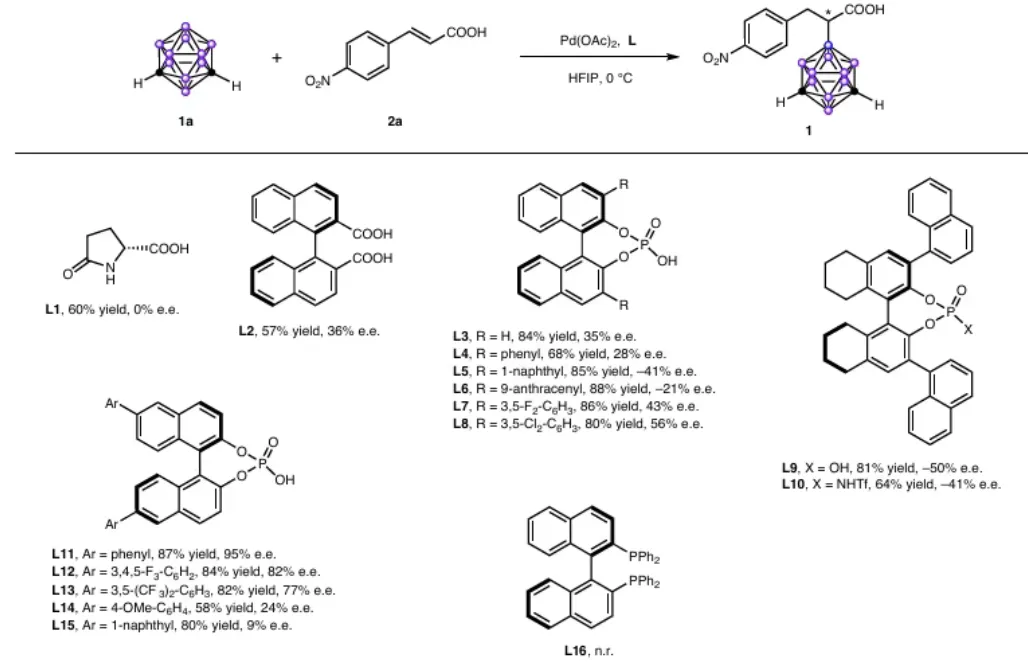
图 2 配体筛选
反应条件:m-碳硼烷(0.105mmol)、4-硝基肉桂酸(0.1mmo)、Pd(Oc) soran(10m 0oC,在空气环境中反应12小时)。通过手性高效液相色谱分析测定所得产物的分离产率和手性选择性。n.r.,表示未发生反应。小结:通过系统筛选手性配体,发现BINOL衍生的6,6'-二苯基手性磷酸(L11)效果最优,在0°C空气中反应12小时,能以87%收率和95%对映选择性获得目标产物。强配位性配体(如BINAP)会完全抑制反应,表明缺电子Pd(II)物种对反应启动至关重要。
图 3|底物拓展
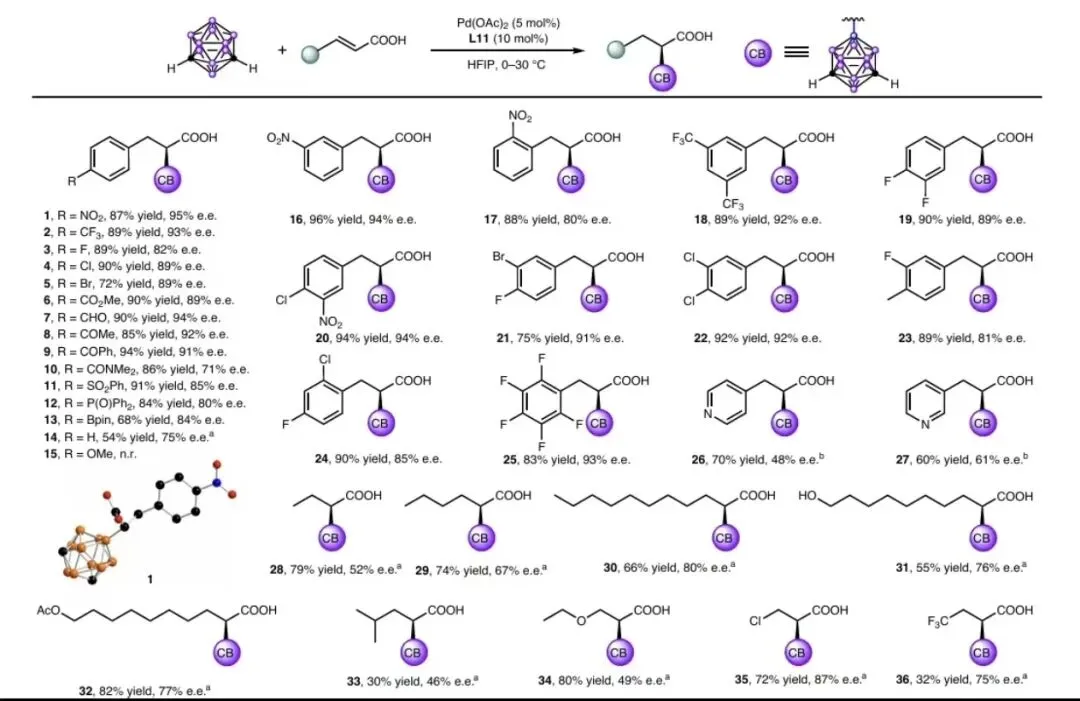
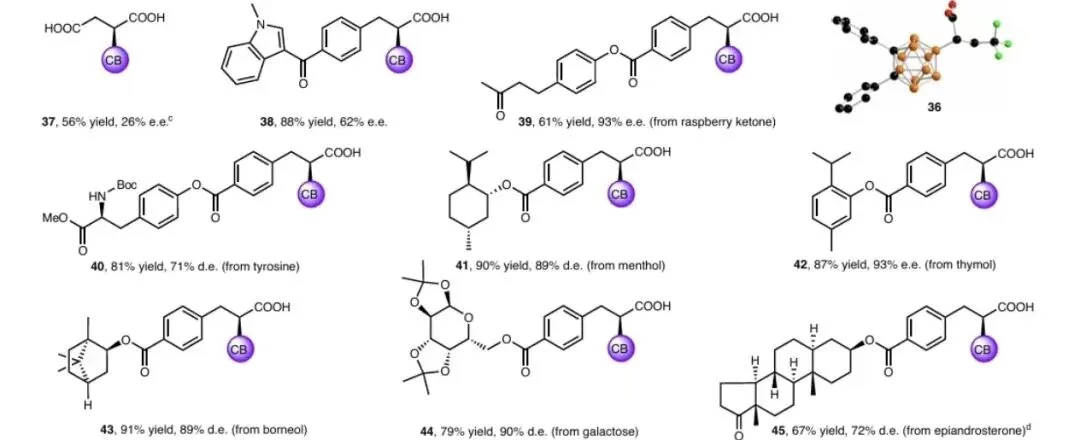
图 3 底物拓展
反应条件: m-碳硼烷(0.105mmol)、a、3-不饱和羧酸 (0.1 mmol) 、 Pd (OAc) (0.005 mmol, 5 mol%) 、 L11 (0.01 mmol,10mol%)置于HFIP(5ml)中,在空气氛围下进行反应,所得产物经分离后,其ee和de值通过手性分析法进行测定HPLC。
小结:对位/间位/邻位取代的肉桂酸衍生物均适用(产物1-25),含吸电子基团时对映选择性更优(最高95% e.e.)。β-烷基取代丙烯酸(28-30)、生物活性分子衍生物(38-45)均可顺利转化,彰显优异官能团耐受性。
图 4|碳硼烷底物
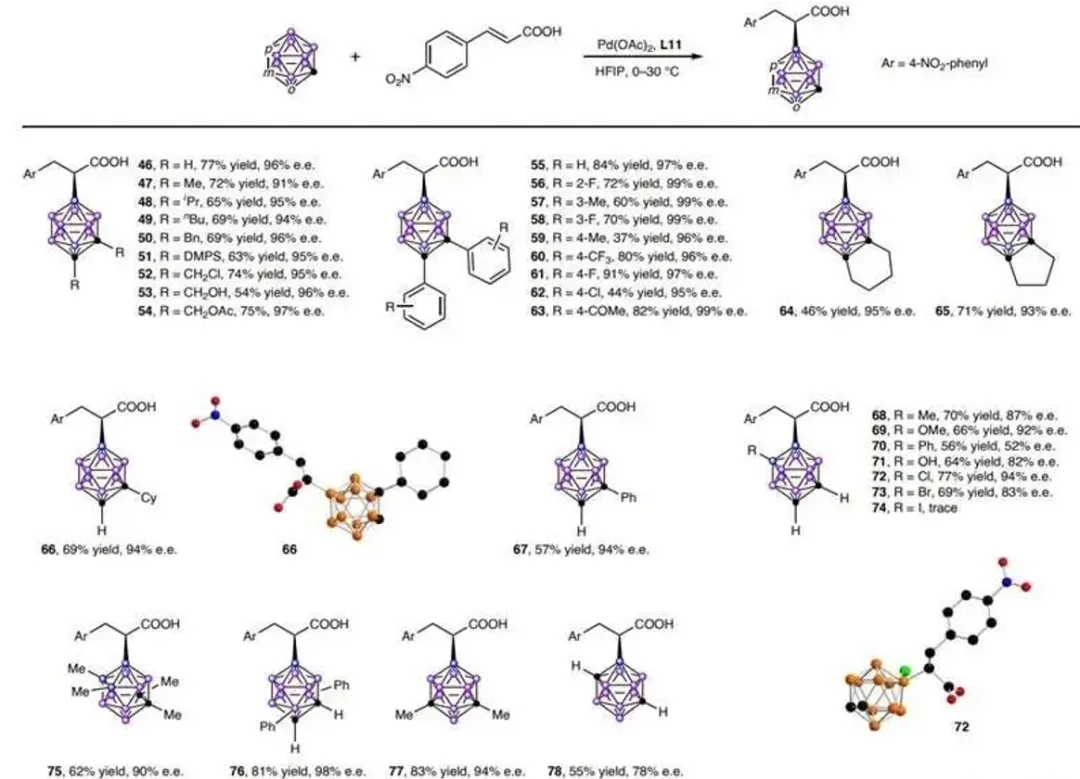
图 4 碳硼烷底物
反应条件: 碳硼烷(0.105 mmo)、4-硝基肉桂酸(0.1mmol)、Pd (OAc)(0.005 mmol, 5mol%)、Ll (0.01mmol,10mol%)置于HFIP(5ml)中,在空气氛围下进行。所分离产物的收率和e.e.值均通过手性高效液相色谱法测定。
小结:邻位/间位/对位碳硼烷均适用,其中邻位碳硼烷的C-芳基取代衍生物可获得99% e.e.(55-63)。B(9)位选择性最高达9:1(B9/B8),单C-取代碳硼烷仍保持高对映选择性(66-67)。
图 5|应用研究
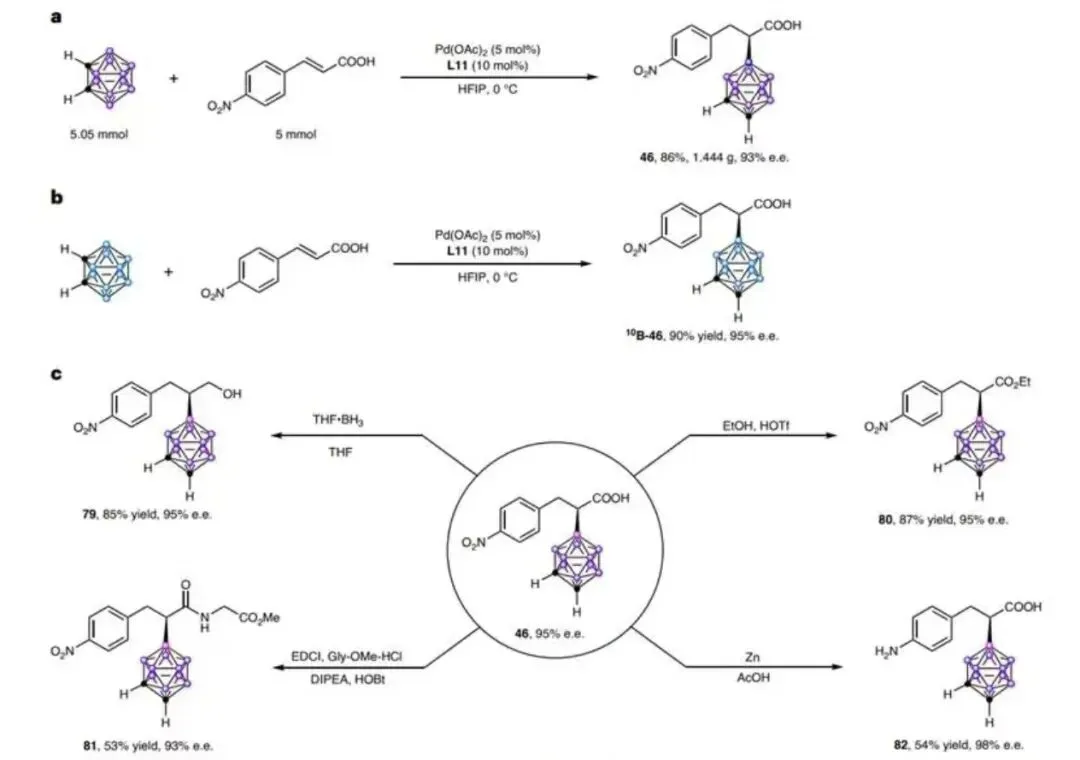
图 5 应用研究
图5a:克级反应; 图5b:标记为2B的产物合成; 图5c,手性a-碳硼烷羧酸的转化
小结:克级反应收率达86%,¹⁰B标记产物可用于硼中子俘获治疗。羧基可转化为酯(80)、酰胺(81)等衍生物,手性中心在酸碱条件下保持稳定,为药物分子修饰提供新途径。
图 7|机理研究
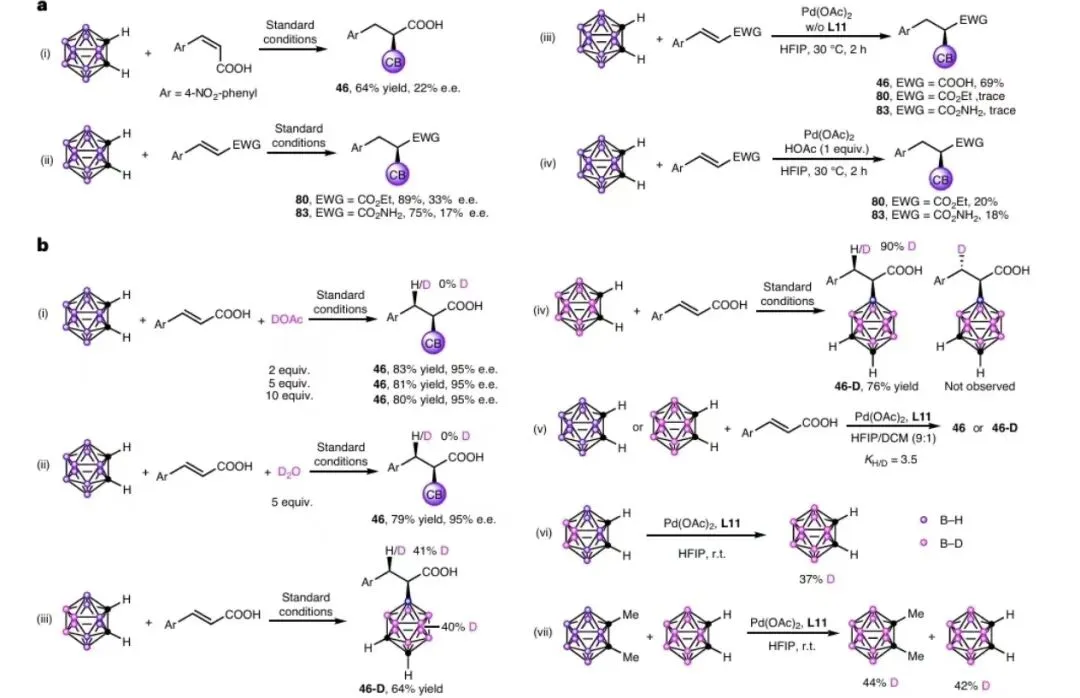
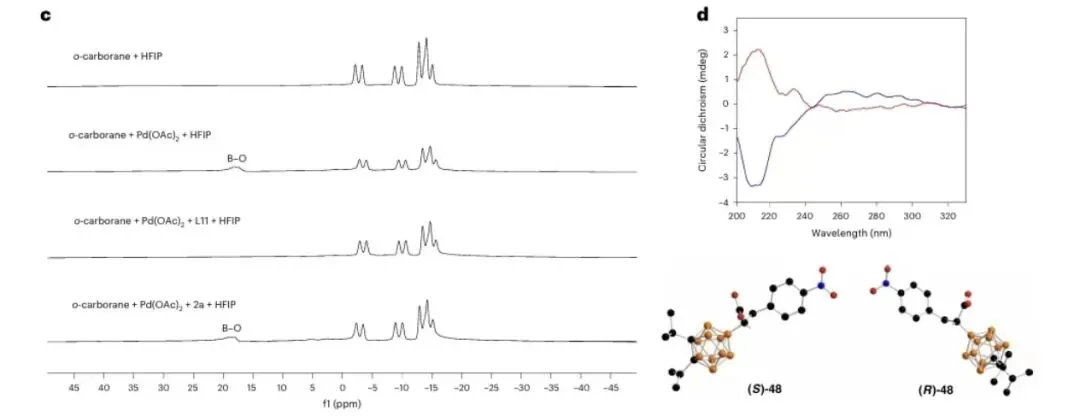
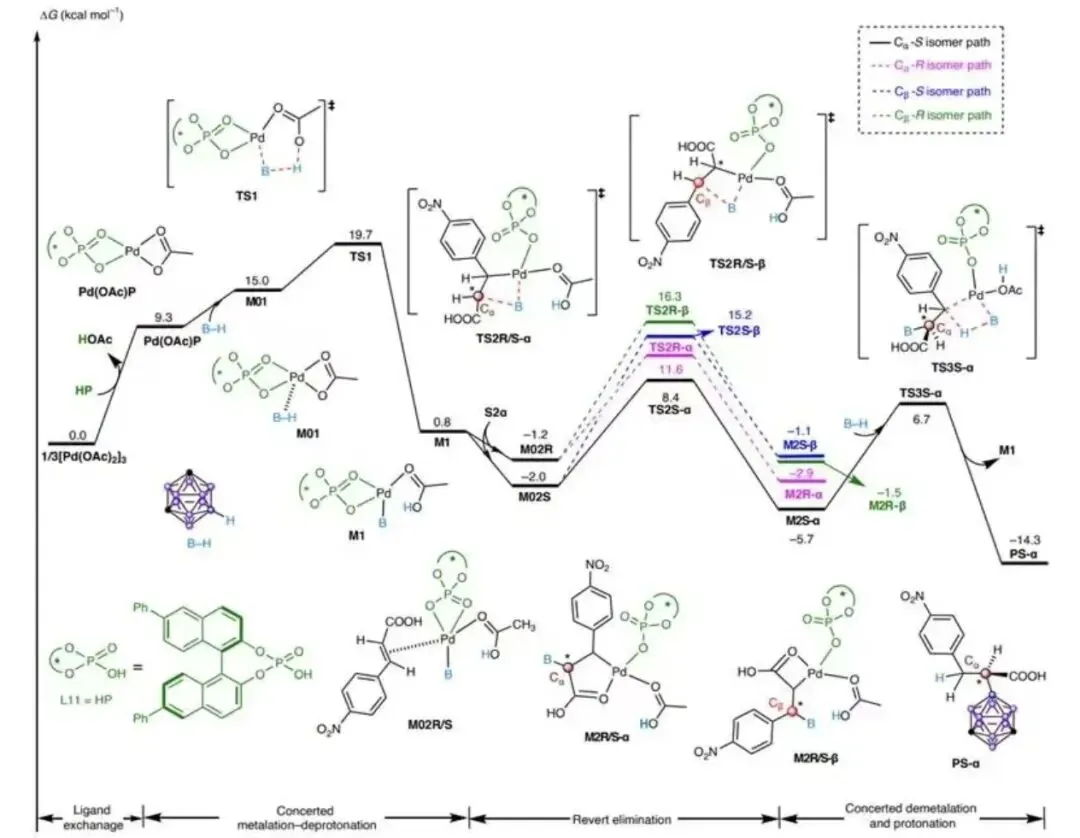
图 7 DFT计算
图7a:对照实验;图7b,氘标记实验。图7c,不同复合物的BNMR图谱。图d,CH,CN(c=0.1mgml1)中(S) -46(蓝色)和(R) -46(红色)的圆二色谱光谱以及(S)-48和(R)-48的分子结构。可能的区域和立体选择性路径的相对吉布斯能量分布情况,采用IEF-PCMary//M06-L/Lanl2DZa/6-31G(d.p)/EF-PCM (a)水平
小结:通过氘代实验与DFT计算,证实反应经历协同金属化-去质子化(CMD)机制:Pd(II)先活化B-H键形成Pd-B中间体,随后烯烃配位并迁移插入形成五元环钯中间体(M2),最终通过协同去金属化/质子化完成α-选择性加成。非共价相互作用分析表明,TS2S-α过渡态中B-H···π等弱相互作用更强,导致S构型路径更优。
以上内容,如有误读和纰漏,敬请指正
杰青联手!华东师范大学关小红/孙远奎&湖南大学王双印,最新Angew:硫掺杂零价铁气凝胶*NO偶联和*H供给协同调控驱动硝酸盐高效电还原成N2 湖南大学「国家杰青」王双印&中南大学任博华&天津大学杨娜,最新Angew:机器学习揭示有机-金属界面“电子海绵”行为促进CO2电还原C2+生成 李灿院士领衔!兰州大学李泽龙,最新Angew:NdO强化IrMnOx双位点协同,酸性OER低Ir长寿命! 韩布兴院士领衔!中科院化学所张裴,最新Angew!温度介导动力学与界面控制以实现浓缩热敏性生物质原料持续电氧化! 孙世刚院士领衔!厦门大学周志有/福州大学郑琦正,最新Angew:Yb掺杂Cu基催化剂!安培级电流密度下跨 pH 范围高效 CO₂ 电还原制 C₂⁺ 包信和院士领衔!复旦大学汪国雄&大连化物所高敦峰/张国辉Angew:离子聚合物驱动反应微环境调控以实现碳酸氢盐介导集成CO₂捕集与电解! 清华大学刘强/山东师范大学王琼,最新Nature Synthesis :电化学钴催化还原偶联实现环丁烷非对映发散合成 南昌航空大学!校史首篇 Science Advances! 师承郑南峰院士,36岁获「国家杰青」黄小青,联手新晋「青年长江」邵琪黄小青,最新JACS:相重排驱动的选择性异质结构构筑 杰青/长江 熊宇杰领衔!中科大龙冉/安徽师范大学熊海龙,最新JACS:Ru/BaTiO₃表面光诱导晶格氧溢流实现高效稳定光热甲烷干重整!

如需转载或合作,请联系我们
联系方式:17739779903(微信同)
联系邮箱:mon@xueyanhui.com
2.催化进展现有综合群、电催化交流群、同步辐射交流群、文献交流互助群、各研究领域群等近20余个,欢迎大家加小编微信,我们会尽快拉您进入对应的群。


