中南大学刘又年&郑州大学王立强ACS Catal.: 基于亲氧金属合金化诱导的–NO₂不对称吸附,打破铂基催化剂选择性合成N-苯基羟胺的标度关系
- 2026-06-12 17:10:52


第一作者:禹敏
通讯作者:刘又年教授,王立强教授
通讯单位:中南大学&郑州大学
论文DOI:10.1021/acscatal.5c07724
在化学工业中,将芳香族硝基化合物高效、高选择性地氢化为N-苯基羟胺(N-PHA)一直是一个极具挑战性的目标。传统的铂(Pt)基催化剂受限于反应中间体之间的线性标度关系,往往陷入“活性”与“选择性”不可兼得的困境,高活性导致过度氢化生成苯胺,而高选择性则常以牺牲反应活性为代价。本文通过在Pt催化剂中引入亲氧金属(OM,如Co、Fe、Ni),成功打破了这一固有的线性标度关系。研究以Pt3Co/AC为模型催化剂,在温和条件(25°C, 2 bar H2)下,实现了硝基苯近乎完全转化(99.9%)和高达96.3%的N-PHA选择性,性能远超传统Pt催化剂。实验和计算结果揭示了Pt–OM位点上独特的“不对称吸附”机制,即在Pt–Co合金位点上,硝基(–NO2)中的一个O原子吸附在Pt位点,另一个O原子吸附在邻近的Co位点,这种独特的吸附模式在空间上解耦了N–O键的活化断裂与不同含N–O中间体的脱附,使得初始N–O键断裂的能垒显著降低,而生成的N-PHA在Co位点上的进一步氢化能垒则相对较高,更易于脱附,从而同时攻克了活性和选择性两大难题。该策略也被证明适用于Pt–Fe和Pt–Ni体系,为理性设计高性能多步加氢催化剂提供了全新的思路。
芳香族硝基化合物的催化氢化是生产苯胺、羟胺、偶氮化合物等一系列重要化工中间体的基石反应。其中,N-PHA作为一种高附加值中间体,是合成特定镇痛药、抗生素、除草剂和杀菌剂等精细化学品的关键前体。然而,N-PHA作为中间体在热力学上并不稳定,极易发生过度氢化,生成更稳定的苯胺。在工业上,为了获得较高的N-PHA收率,常常不得不使用昂贵且易产生副产物的强还原剂(如水合肼),并在极低的温度(如0°C)下进行操作,增加了成本和工艺复杂性。使用清洁、廉价的H2作为还原剂是更理想的选择。Pt等贵金属催化剂能在温和条件下高效活化H2,因此备受关注。而Pt表面通过d-π轨道重叠对芳香环具有强烈吸附作用,硝基苯倾向于硝基的对称性双齿平行吸附,这种吸附模式有利于硝基的加氢,但对生成的中间体同样具有强吸附,迫使其持续加氢直至N–O键完全断裂生成苯胺。即,反应物(硝基)和含有N–O键的中间体存在固有的“线性标度关系”。此前,利用配体修饰或载体工程来削弱芳香环的吸附,是提高N-PHA选择性的常见策略。但这往往导致对硝基本身的吸附也变弱,使得第一步N–O键断裂(决速步骤)的活化变得困难,使得催化活性显著下降。这种“活性-选择性”的权衡,其根源正是不同含N–O中间体在单一金属位点上的吸附能之间存在的线性关联。因此,要真正实现突破,必须找到一种策略能够差异化地调控对反应物和不同中间体的吸附强度,即打破线性标度关系。
1. 打破固有线性标度关系:通过Pt与亲氧金属合金化,首次在该反应中同时实现高活性与高选择性,突破了传统催化剂的设计瓶颈。
2. 揭示“不对称吸附”新机制:实验与理论计算结合,阐明Pt–Co双位点诱导硝基不对称吸附的机理,并实现反应物活化与产物脱附的功能分区。
3. 性能卓越且条件温和:Pt3Co/AC在近室温、低氢压下表现出较好的转化率与选择性,且具备优异的底物普适性和循环稳定性。
4. 策略具备普适性:PtFe/AC与PtNi/AC同样表现出显著优于Pt/AC的性能,证明亲氧金属合金化是普适性策略。
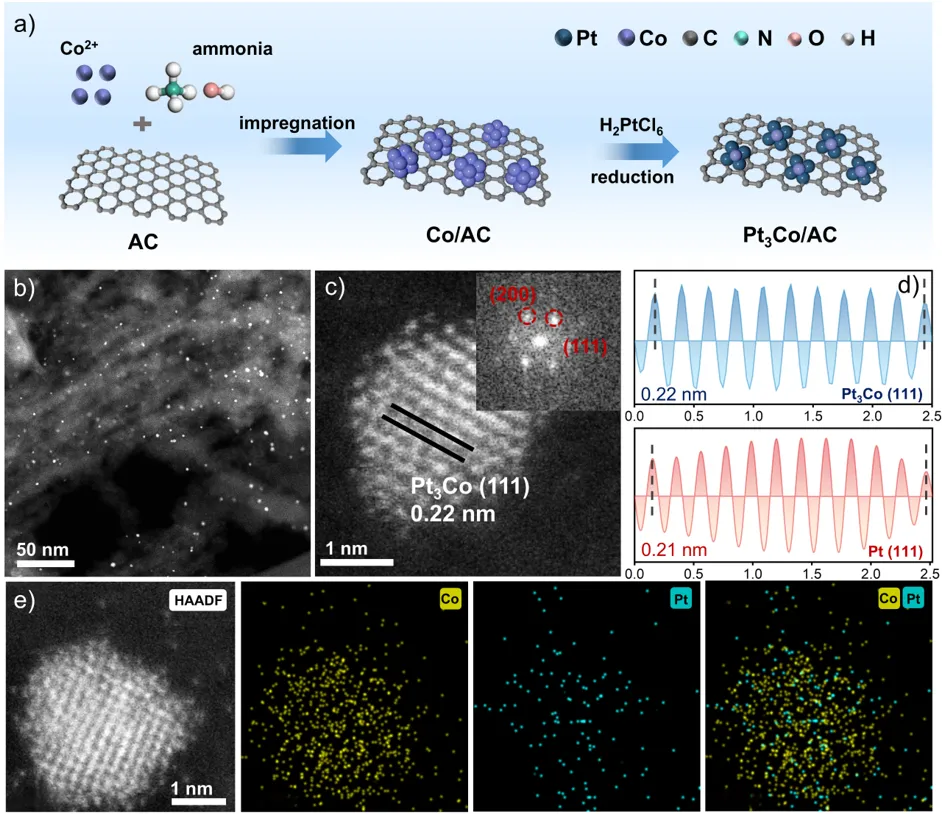
如图所示,采用顺序沉积-沉淀法,经过煅烧和还原合成了活性炭(AC)负载的Pt3Co合金纳米催化剂。高分辨透射电镜(HRTEM)和球差校正HAADF-STEM成像显示,纳米颗粒均匀分散在碳载体上,其平均尺寸为2.7 nm,且Pt和Co元素在纳米颗粒内均匀分布,验证了Pt–Co合金结构的形成。
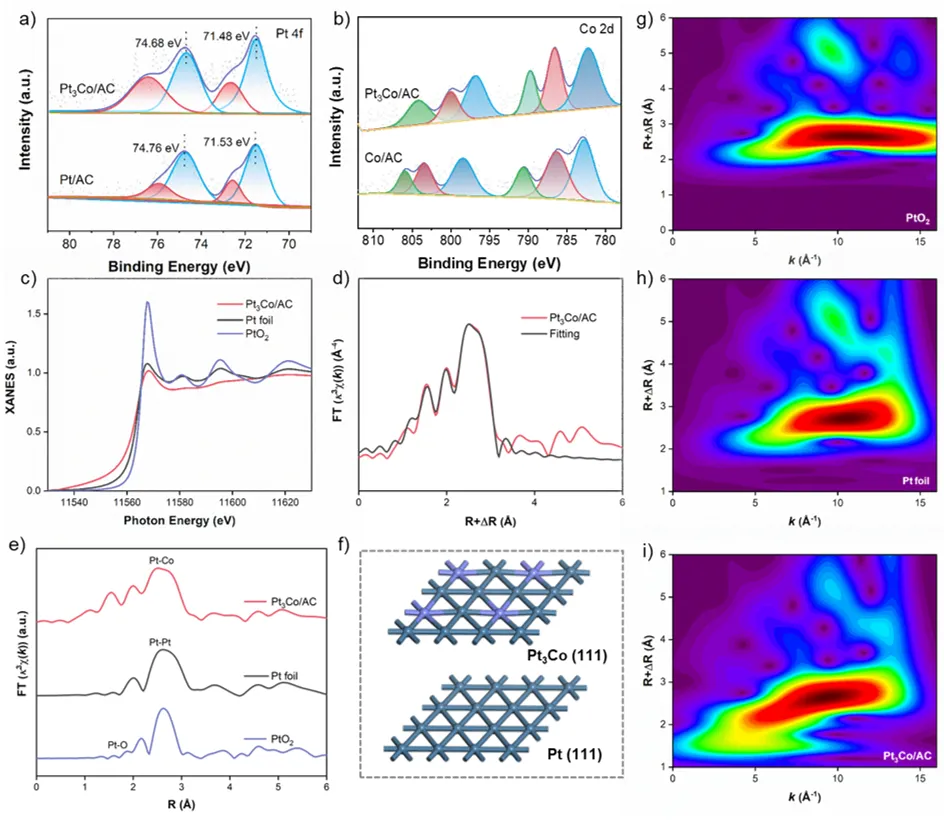
通过XPS和EXAFS表征,进一步明晰催化剂的配位环境和电子结构。XPS分析表明电子从Co向Pt转移,改变了Pt的电子结构,削弱了其d轨道与苯环π电子的耦合能力。EXAFS进一步证实了Pt–Co配位键的形成,并揭示Pt与Co配位形成Pt3Co结构。
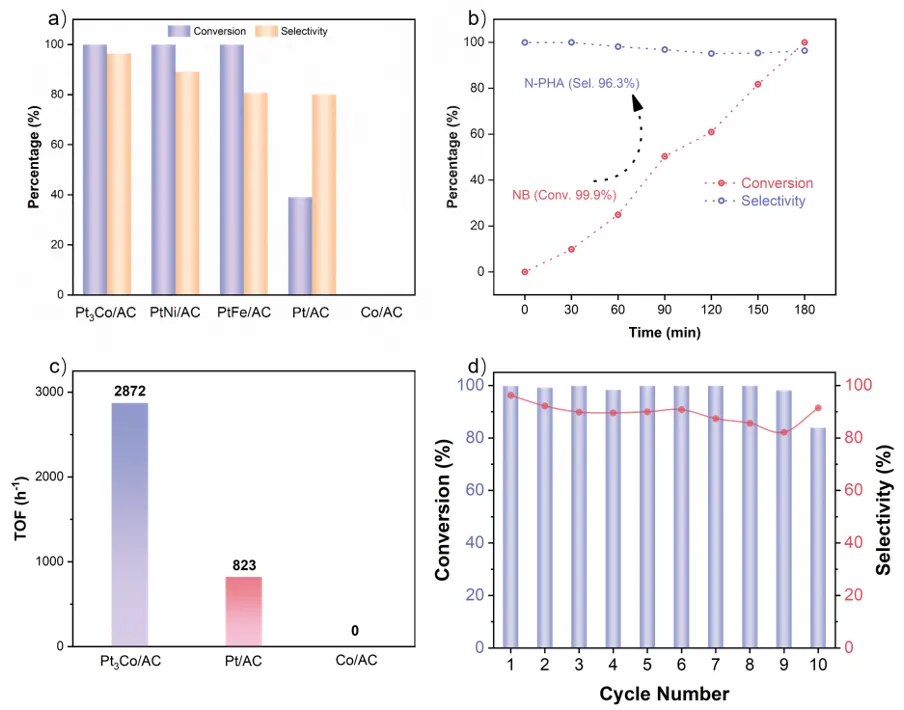
以硝基苯加氢为模型反应,Pt3Co/AC表现出较好的催化性能。在25°C、2 bar H2的温和条件下,3 h内实现了99.9%的硝基苯转化率和96.3%的N-PHA选择性,显著优于Pt/AC、商业Pt/C等催化剂。此外,测试了不同催化剂的本征活性,Pt3Co/AC催化剂的TOF值高达4201.5 h-1,比Pt/AC(3372.8 h-1)提升了25%,表明Co的引入显著提高了选择性和本征活性。同时,催化剂展现出优异的底物普适性,并在重复使用10次后,活性和选择性均未发生明显衰减,结构稳定性良好。
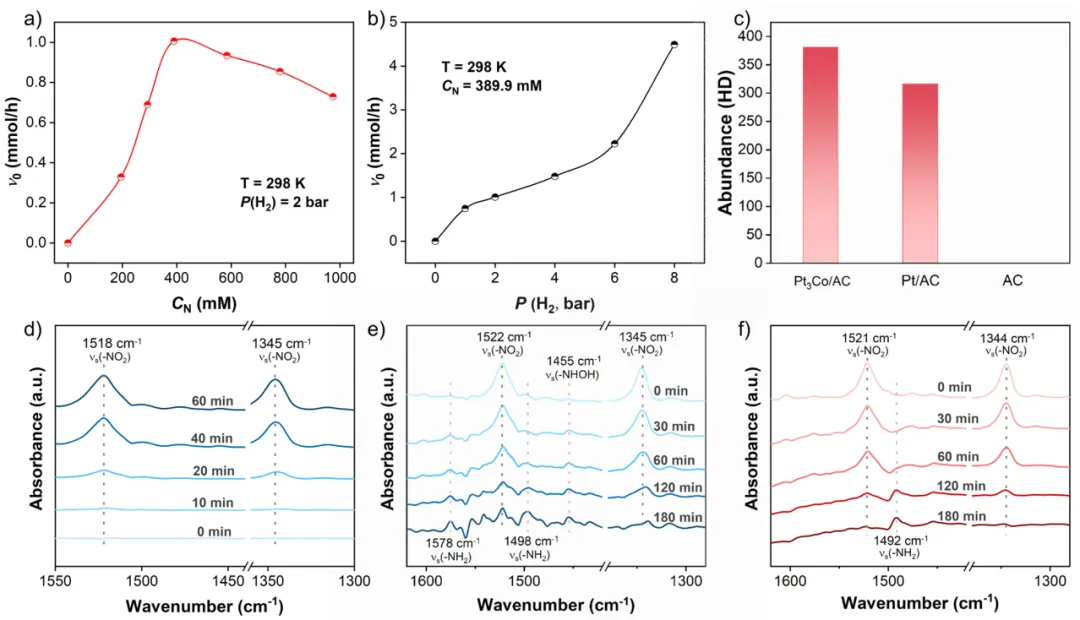
利用动力学实验、原位光谱和密度泛函理论(DFT)计算进一步揭示催化机制。H2-TPD和H-D交换实验表明,Pt3Co/AC具有更强的H2吸附和解离能力,为加氢反应提供充足的表面氢物种。原位漫反射红外光谱(DRIFTS)揭示了不同催化剂上的吸附构型。在Pt/AC上,硝基苯的吸附以苯环的平行吸附为主。而在Pt3Co/AC仅观察到–NO2对应的特征峰,说明Co的引入会显著削弱催化剂对苯环的吸附,同时增强了对–NO2基团的定向吸附。通过CO选择性毒化Pt位点后,Pt3Co/AC对硝基苯的吸附几乎不受影响,而Pt/AC的吸附显著减弱。综合表明在Pt3Co/AC催化剂上,金属Co是吸附和锚定–NO2基团的关键位点。
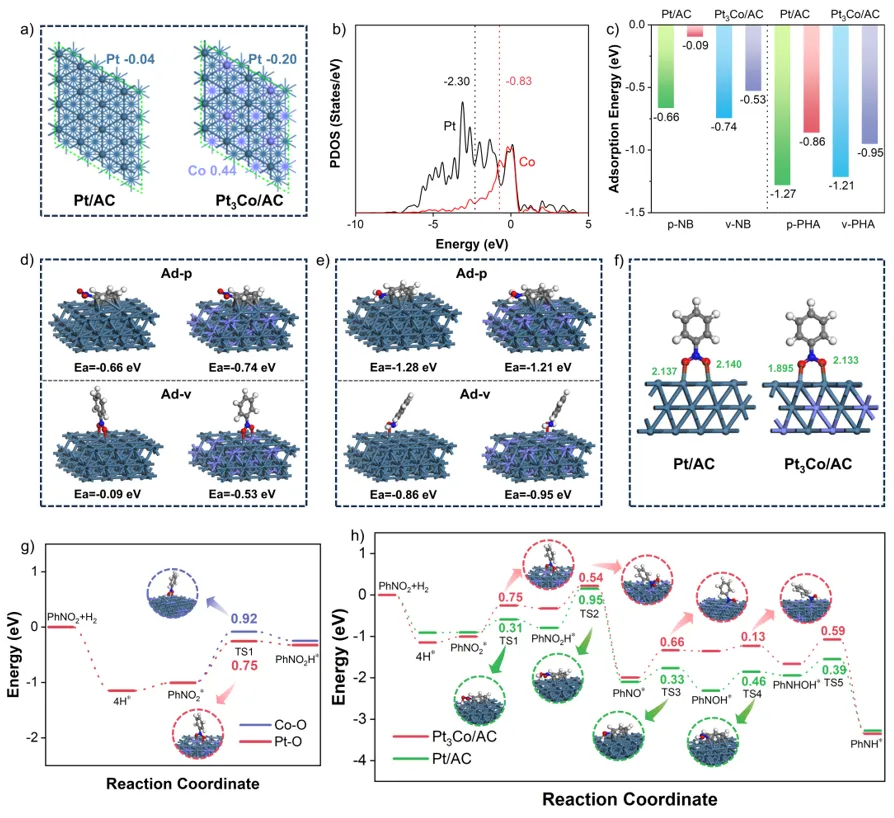
DFT计算表明,由于Co的亲氧性,Pt3Co(111)表面显著增强了对硝基苯的垂直吸附。–NO2的两个O原子分别与Pt和Co原子成键,Co–O键比Pt–O键更短,形成不对称的吸附构型。这种不对称性导致功能的精准空间分区,Pt位点作为初始活化中心,第一个N–O键(Pt–O)断裂的能垒较低,显著降低了氢化过程中的决速步能垒(RDS)。Co位点作为后续加氢位点与脱附调节中心,其强亲氧性能够稳固中间体,但生成的N-PHA在该位点上进一步氢化的能垒远高于其直接脱附的能垒。因此,在Pt3Co(111)上反应路径被重构,整个过程将反应物的活化与目标产物的脱附在空间和功能上解耦,从而同时降低了RDS并抑制了过度加氢。
制备了PtFe/AC和PtNi/AC催化剂,通过结构表征证实它们也形成了合金。在相同的温和条件下均表现出较好的催化性能,验证了亲氧金属合金化策略的广泛适用性。
总之,将Pt与亲氧金属(OM=Co、Fe、Ni)合金化能够打破硝基芳烃催化氢化合成N-PHA过程中吸附标度关系所施加的活性-选择性限制。与Pt/AC或商用Pt/C相比,Pt3Co/AC表现出优异的N-PHA选择性。实验和理论计算表明,与亲氧金属合金化会在Pt–OM位点诱导–NO2的不对称吸附。这种独特的吸附结构使硝基苯氢化过程中不同的N–O键在不同的位置发生断裂,有效地打破了氢化过程中固有的线性标度关系。该策略不仅降低了在氢化过程中的能量势垒,而且抑制了N-PHA的进一步氢化,从而克服了催化氢化中的活性-选择性权衡。这项研究不仅开发了一种高性能的N-PHA合成催化剂,更为设计多步串联反应催化剂提供了全新的方法论。通过构建具有差异化吸附特性的相邻活性位点,可以精准调控复杂反应网络中特定步骤的动力学,从而实现对目标产物的高选择性合成。这一理念有望扩展到其他涉及多个中间体、受标度关系制约的重要催化反应中,为下一代高性能催化剂的开发开辟了新的道路。
Yu M; Ouyang, D; Zhao X; Ren Z; Yao F; Wang L; Liu Y-N. Breaking Scaling Relations for Selective N‑Phenylhydroxylamine Synthesis on Platinum Catalysts via Oxophilic Metal Alloying‑Induced Asymmetric Adsorption of –NO2. ACS Catal. DOI: 10.1021/acscatal.5c07724
刘又年,中南大学化学化工学院教授,博导。湖南省微纳材料界面科学重点实验室主任。中国化工学会理事、湖南省化学化工学会副理事长。主持国家重大研发计划课题、国家自然科学基金重点项目、面上项目等10余项。主要从事纳米催化剂的设计及其在光电热催化及生物医药中的应用研究。在JACS, Adv. Mater., Angew. Chem. Int. Ed., PNAS, Sci. Adv., Matter, ACS Catal., AIChE J等刊物发表学术论文200余篇,论文引用22000余次,H-index 77,授权国家发明专利20余项,获得省部级自然科学奖、科技进步奖等5项。
王立强,郑州大学材料科学与工程学院教授,博士生导师,河南省优秀青年基金获得者。主要多相催化的实验与理论计算研究,聚焦碳基催化剂的设计、构筑及应用;硝基化合物的选择性加氢及烷烃脱氢反应等方面的过程控制、构效关系研究以及新路线开发。目前已在J. Am. Chem. Soc., Angew. Chem. Int. Ed., Adv. Mater., ACS Catal., J. Catal等期刊发表论文70余篇,被引近5000余次,获授权发明专利10件。先后承担国家自然科学基金重点项目、面上项目和青年项目等纵向课题以及河南省重大科技攻关、郑州市重大协同专项等横向课题。
版权声明
测试表征+计算+绘图

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 春风马蹄疾,桃李贺新春!郑州市第一二九中学全体教师送祝福啦!
- 超长春节假期,郑州金水奉上“文旅盛宴”!最全打卡攻略来啦→
- 郑州大学文学院简介,始建于1958年,有教职工114人,有教授25人,副教授32人,有中国语言文学一级博士学位授权点和一级硕士学位授权点
- 2026新春怎么玩?郑州航空港三大宝藏园区嗨玩整个新年,内附节目单
- 扮靓新春系列|马年春节不停歇 郑州城管全力护航城市平稳运行
- 3月液针刀中医微创疼痛实战研修班—(郑州站)火热报名中!
- 郑州西京白癜风医院寒假快速祛白计划仍在继续——七大检查仅177.5元+308激光最高报40%!
- 郑州大学、湖南科技大学等5所院校发布2026年体育单招简章
- 郑州北龙湖湿地公园
- 郑州蓝天健康体检服务院2026年会庆典: 为梦想再起航,共绘健康新篇
