陕西科技大学刘昭铁/何珍红&郑州大学杨得鑫,最新Angew!基于Cu-MOF设计多价态Cu催化剂实现高性能C₂电合成!曹会会一作
电催化CO₂还原反应(ECO₂RR)是将CO₂转化为有价值化学品,特别是C₂₊产物的重要过程,然而实现对C₂₊产物的高选择性仍具挑战。在目前已探索的催化剂中,具有多价Cu的Cu基催化剂被广泛用于此过程。然而,Cu基催化剂的精确调控,特别是多价Cu物种的稳定,仍是提高C₂₊产物选择性的难题。
2026年03月02日,陕西科技大学刘昭铁、何珍红团队在Angewandte Chemie International Edition期刊发表题为“Engineering Multivalent Copper Catalysts From Cu-MOF Towards High-Performance C2 Electrosynthesis”的研究论文,团队成员曹会会为论文第一作者,刘昭铁、郑州大学杨得鑫、何珍红为论文共同通讯作者。
第一作者:曹会会
通讯作者:刘昭铁、杨得鑫、何珍红
通讯单位:陕西科技大学、郑州大学
论文DOI:10.1002/anie.202524816
该研究报道了一种高效Cu基催化剂(命名为Cu-MOF/CuₓO/CF-40),该催化剂包含在Cu箔(CF)上生成的对苯二甲酸Cu(PTA)(Cu-MOF)以及主要源于Cu-MOF的CuₓO(Cu₂₊₁O和CuO),用于实现ECO₂RR制C₂化学品。该催化剂在0.1 M KCl(H型电解池)中,于-1.1 V(vs. RHE)电位下,实现了高达91.8%的总C₂法拉第效率(FE C₂)和34.6 mA·cm⁻²的总电流密度。具有多价Cu特性的Cu₂₊₁O和CuO物种能够吸附并活化CO₂,并提高催化剂的导电性。Cu-MOF则稳定了催化剂的多价态特性和长期性能。原位ATR-FTIR和DFT计算确定了*OCCOH是关键C–C偶联中间体。该研究为设计高效、稳定Cu基催化剂提供了一种合理策略,并展示了一种构建多价Cu位点以实现CO₂选择性向C₂转化的单一来源路线。
过量CO₂排放对全球可持续发展和人类安全构成严重威胁。为此,CO₂的直接转化吸引了大量的研究关注。由可再生能源驱动的电催化CO₂还原反应(ECO₂RR)为将CO₂转化为有价值的化学品和燃料提供了一条有前景的途径。在多种ECO₂RR产物中,多碳(C₂₊)化合物因其比C₁产物更高的能量密度和更广泛的应用潜力,从商业角度来看尤为理想。然而,C₂₊产物的生成涉及复杂的多质子、多电子转移步骤,导致缓慢C–C偶联动力学,严重制约了这些产物的活性和选择性。因此,设计高效、高活性且稳定的电催化剂对于在ECO₂RR中实现高活性和高选择性的C₂₊产物至关重要。
Cu基材料因其独特的电子性质有利于C–C偶联,被广泛认为是ECO₂RR中生产C₂₊化合物的高效催化剂。迄今为止,存在于多种氧化态中的多价Cu物种作为生成C₂₊产物的活性位点已被广泛研究。然而,这些物种在阴极条件下不稳定,易被还原为金属Cu(Cu⁰),这限制了其实际应用。此外,精确调控其组成和局域微环境以提高C₂₊选择性仍然是一项相当大的挑战。因此,稳定混合价态Cu物种和调控其催化微环境的困难构成了一个主要障碍,阻碍了Cu基催化剂在ECO₂RR中的更广泛应用。
Cu基MOF材料为ECO₂RR反应提供了丰富的多孔结构。大量配体可以在应用过程中稳定Cu物种。其明确的多孔结构和高比表面积有利于传质并暴露出丰富的活性位点,促进CO₂的吸附和活化。Cu中心的可调配位环境允许对催化选择性进行精确控制,特别是对于高价值C₂₊产物的生成。此外,Cu-MOF固有的结构灵活性赋予其良好的复合构建兼容性,当与其他功能组分(如金属氧化物、导电基底)集成时,能够产生协同效应,以增强导电性和催化稳定性。例如,Sun等人通过一步溶剂热法将CuO掺入Cu-MOF中,在10小时内实现了超过45%的C₂H₄法拉第效率。此外,将Cu箔与特定位置的MOF耦合,增强了Cu活性位点并抑制了CH₄生成,最终实现了48.6%的C₂H₄法拉第效率。Yang等人将Cu基MOF电化学还原为介孔Cu纳米带,实现了82.3%的FEC₂。尽管取得了这些进展,Cu-MOF催化剂在ECO₂RR过程中仍存在耐久性和稳定性差的问题,这主要是由于Cu-配体配位键较弱所致。为应对这一关键挑战,Zhang等人报道了一种柔性Cu(I)-三唑骨架,通过改变配体侧基的尺寸,实现了对C₂H₄/CH₄选择性的逐步调控。经过长期电催化后,该骨架保持了其原始结构和形貌,未形成Cu基无机物种。Zhang等人还开发了一种缺陷工程化的Cu-MOF(UC-Cu-BTEC),实现了77.2%的C₂产物法拉第效率,并保持了超过10小时的稳定性能。
受这些进展的启发,该研究采用Cu箔(CF)作为Cu源以生成Cu-MOF,并进一步衍生得到具有多价Cu物种的CuₓO(记为Cu-MOF/CuₓO/CF-t),用于高效ECO₂RR制C₂产物。最优Cu-MOF/CuₓO/CF-40催化剂在H型电解池中,0.1 M KCl电解液内,实现了高达91.8%的FE C₂和34.6 mA·cm⁻²的总电流密度,并具有超过40小时的良好稳定性。与传统的CuO不同,Cu²⁺¹O相具有富含缺陷的阶梯状结构,含有混合价态的Cu离子,提供了高密度的活性位点。其丰富的金属缺陷和氧空位在调控Cu的电子结构、促进CO₂吸附和活化方面起着关键作用。以Cu₂₊₁O和CuO形式存在的多价Cu物种的同时存在,稳定了化学吸附的CO₂中间体。Cu-MOF组分增加了催化剂的表面积和孔体积,有利于传质和离子扩散,并稳定了多价Cu,而Cu₂₊₁O和CuO相则增强了导电性并为反应提供了活性位点。该研究为将CO₂转化为C₂产物提供了一种有效的策略,并为设计具有多价态、丰富缺陷和多样化活性位点的高效Cu基催化剂提供了宝贵见解。
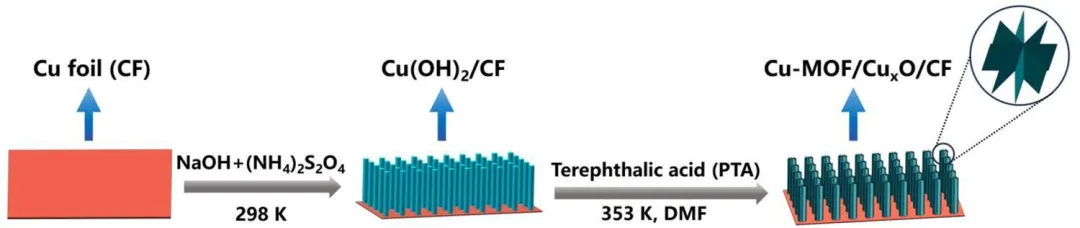
示意图1 Cu-MOF/CuₓO/CF-t电极的合成步骤。
图1 Cu-MOF/CuₓO/CF-40的 (a–c) TEM图像,(d) SAED测试区域(I, II),(e, f) 高分辨率TEM (HRTEM) 图像,(g) SAED图谱,(h) HAADF图像,以及 (i–m) 元素分布图。
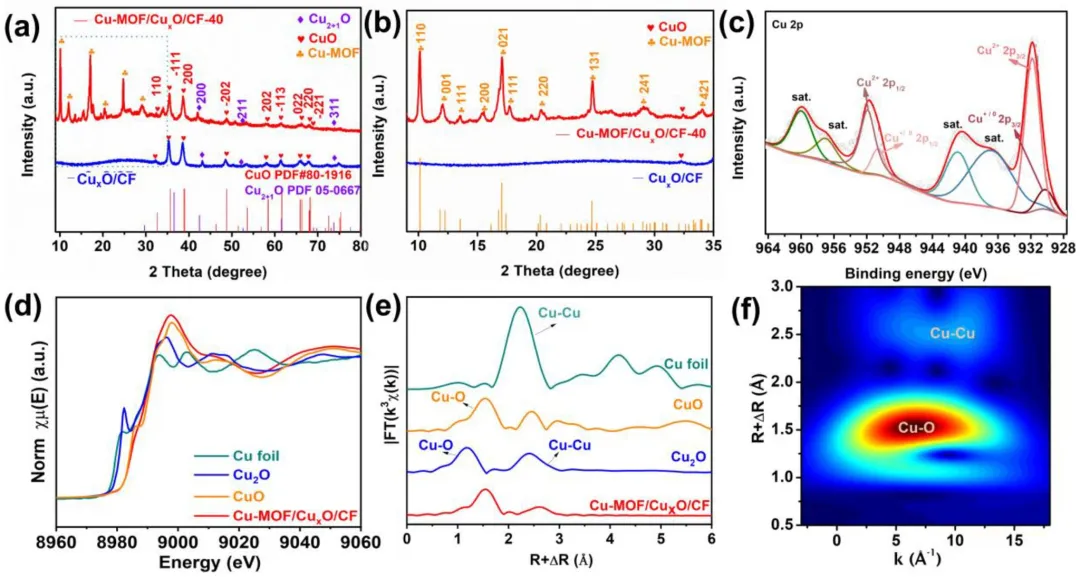
图2 (a) Cu-MOF/CuₓO/CF-40的XRD图谱及对应的参考PDF卡片。(b) 图 (a) 中5–35°范围内的放大XRD图谱。(c) Cu-MOF/CuₓO/CF-40的Cu 2p XPS能谱。(d) Cu箔、Cu₂O、CuO和Cu-MOF/CuₓO/CF-40的Cu K-edge XANES图谱。(e) Cu箔、Cu₂O、CuO和Cu-MOF/CuₓO/CF-40的傅里叶变换k³权重χ(k) EXAFS图谱。(f) Cu-MOF/CuₓO/CF-40的Cu K-edge EXAFS数据的小波变换。
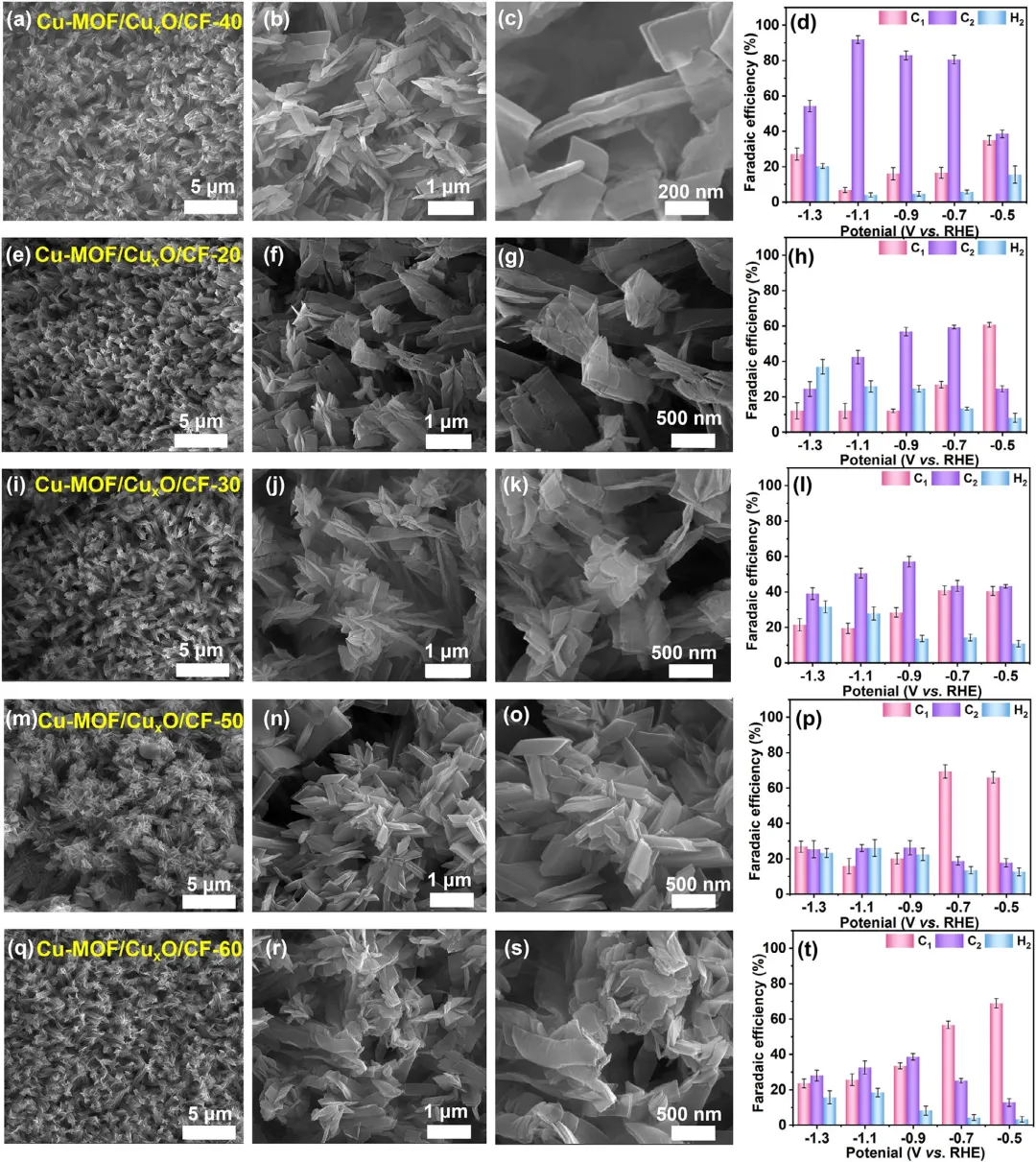
图3 通过碱性溶液浸泡 (a–c) 40、(e–g) 20、(i–k) 30、(m–o) 50 和 (q–s) 60 min合成的Cu-MOF/CuₓO/CF的SEM图像。在CO₂饱和的0.1 M KCl电解液中,通过碱性溶液浸泡40 (d)、20 (h)、30 (l)、50 (p) 和 60 (t) min合成的Cu-MOF/CuₓO/CF-t催化剂在不同外加电位下的ECO₂RR催化性能。误差棒表示三次独立实验的平均值±标准偏差。
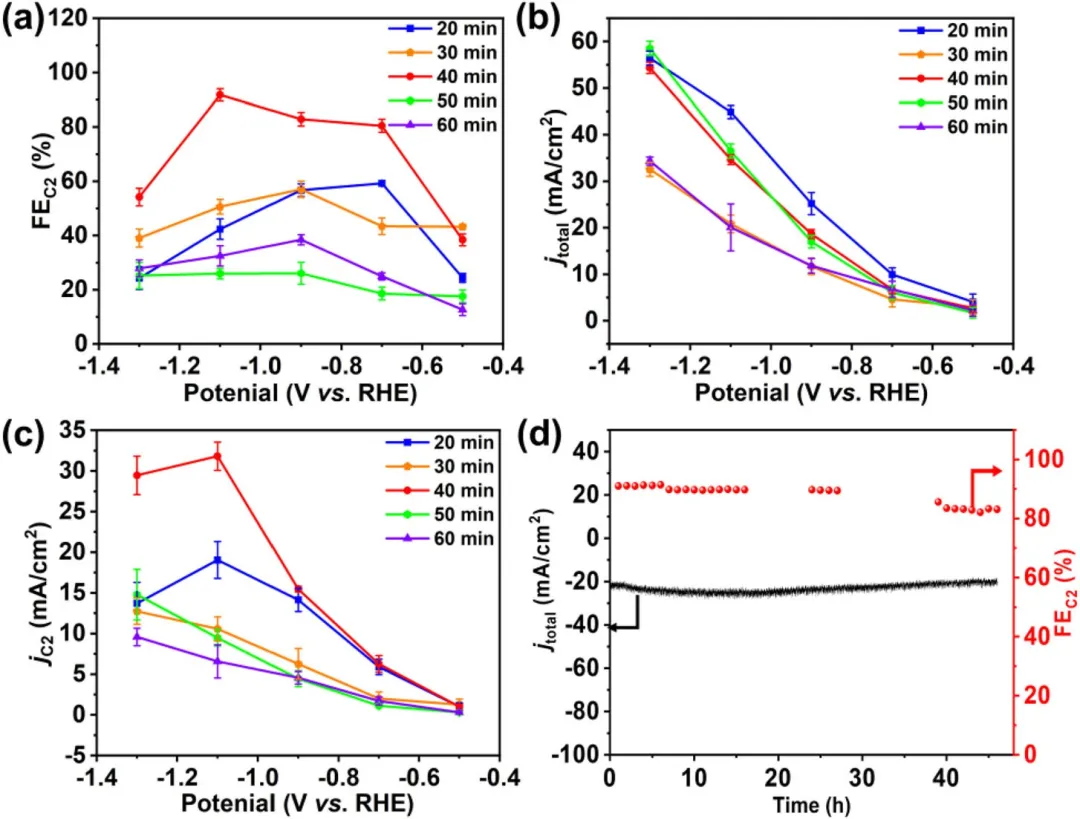
图4 不同浸泡时间(t = 20、30、40、50 和 60 min)制备的Cu-MOF/CuₓO/CF-t的 (a) C₂产物总FE,(b) 总电流密度,(c) C₂产物分电流密度(分电流密度均归一化至CF的几何表面积),以及 (d) Cu-MOF/CuₓO/CF-40催化剂在CO₂饱和0.1 M KCl电解液中于-1.1 V (vs. RHE) 下的稳定性。误差棒表示三次独立实验的平均值±标准偏差。
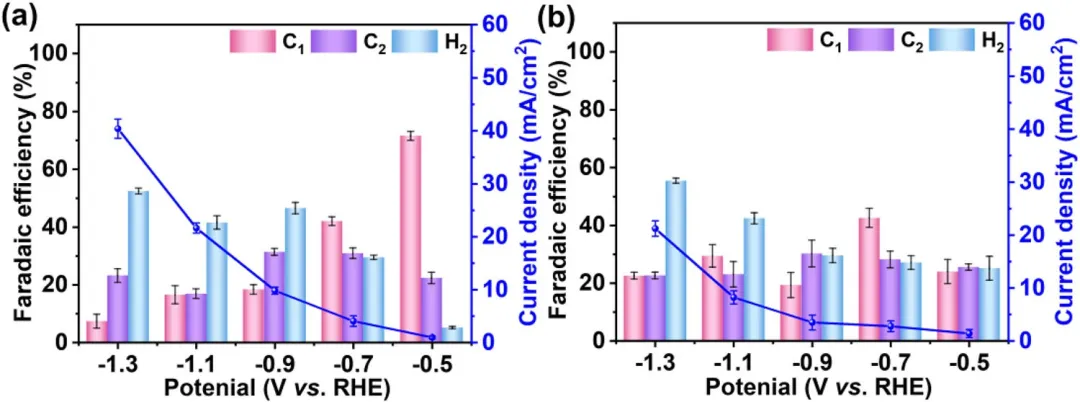
图5 (a) CuₓO/CF和 (b) CF样品在CO₂饱和的0.1 M KCl电解液中,不同电位下的ECO₂RR催化性能。误差棒表示三次独立实验的平均值±标准偏差。
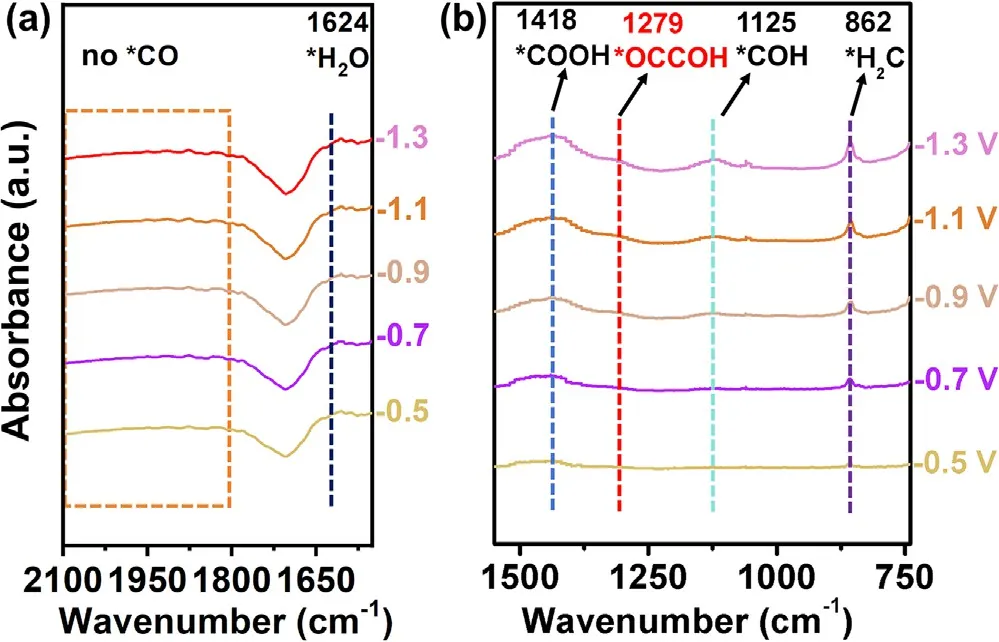
图6 Cu-MOF/CuₓO/CF-40催化剂在-0.5~ -1.3 V (vs RHE) 不同电位下ECO₂RR的原位ATR-FTIR光谱。
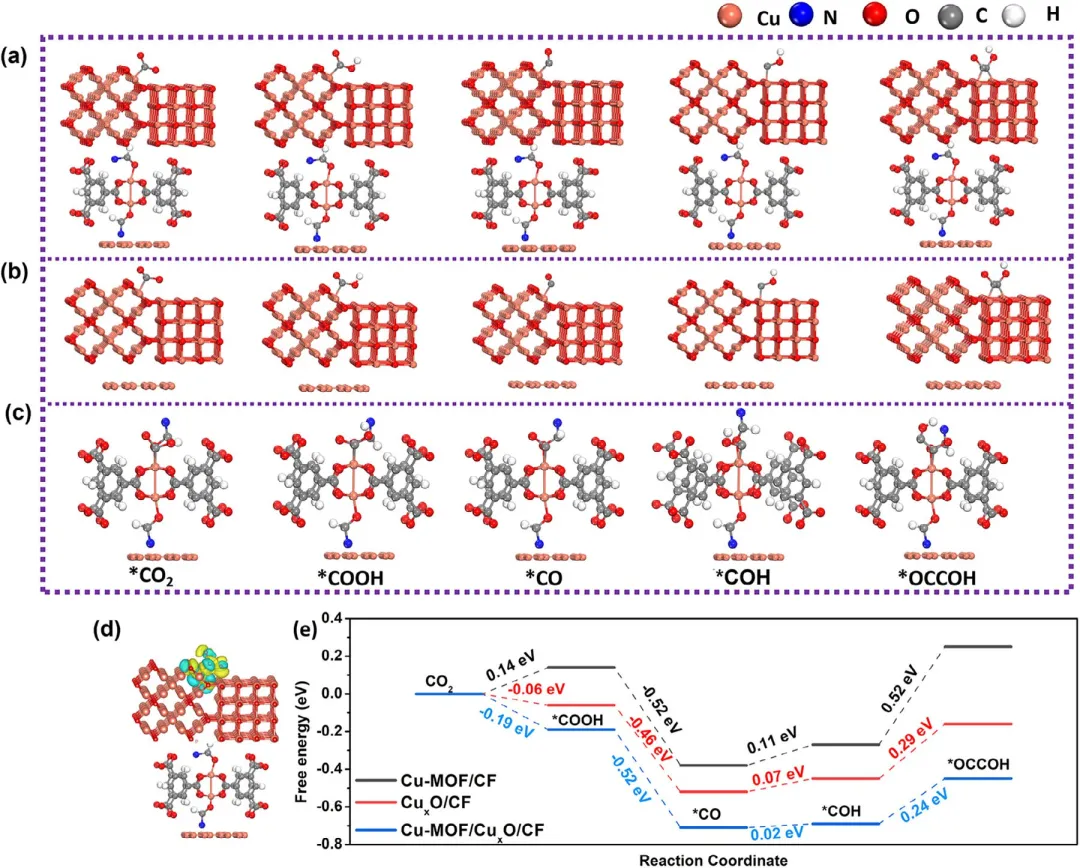
图7 ECO₂RR制C₂产物的DFT计算。(a) Cu-MOF/CuₓO/CF-40,(b) CuₓO/CF,和 (c) Cu-MOF/CF的催化剂结构。(d) CO₂在Cu-MOF/CuₓO/CF-40表面吸附的差分电子密度图,其中黄色和蓝色区域分别代表电子聚集和耗尽。(e) ECO₂RR在Cu-MOF/CuₓO/CF-40(蓝色)、CuₓO/CF(红色)和 Cu-MOF/CF(黑色)上生成 C₂产物的反应路径的DFT计算。
总之,该研究通过两步原位合成法成功制备了Cu-MOF/CuₓO/CF-40催化剂,并评估了其在电催化还原CO₂制C₂产物中的性能。该催化剂具有由片状亚单元组成的独特棒状纳米阵列结构,该结构是通过在Cu箔基底上原位生长Cu-MOF、Cu₂₊₁O和CuO相而形成的。在H型电解池中,于-1.1 V(vs. RHE)电位下,该催化剂对C₂产物实现了高达91.8%的法拉第效率,同时总电流密度达到34.6 mA·cm⁻²,并在连续电解40小时内表现出显著的操作稳定性。结合原位ATR-FTIR光谱和DFT计算证实,增强的C₂反应路径经由关键中间体(*CO和*COH),最终通过*OCCOH的形成完成关键的C–C偶联步骤。该研究不仅阐明了一条高效CO₂向C₂转化催化路径,而且为设计稳定且高活性的多价Cu基催化剂提供了重要见解,突显了MOF衍生结构在先进电催化中的协同潜力。