钠离子电池通过解决资源稀缺性和安全性问题,在可持续能源存储方面具有重要前景,提升上限截止电压对于钠离子电池充分发挥电极容量并缩小其与商用锂离子电池之间的能量密度差距至关重要。然而,常规电解液在高脱钠态正极表面表现出电化学不稳定性,遭受强亲电攻击并持续分解,导致形成富含低聚物的界面相和快速容量衰减。
2026年05月06日,郑州大学陈卫华团队在Nature Communications期刊发表题为“Dual-domain solvent-locked electrolyte enabled durable 4.5 V-class sodium batteries”的研究论文,团队成员张继雨、唐国串为论文共同第一作者,陈卫华为论文通讯作者。
第一作者:张继雨、唐国串
通讯作者:陈卫华
通讯单位:郑州大学
论文DOI:10.1038/s41467-026-72849-z
该研究设计了一种溶剂锁定型碳酸酯基电解液,形成了电化学稳定的溶剂增强型溶剂化结构以及在正极上由阴离子富集界面屏蔽层衍生的富硼化物/氟化物的界面相,从而抑制了漏电流和寄生反应。该定制电解液与商用氧化物和聚阴离子正极表现出良好相容性。组装的Na|Na₂.₂₆Fe₁.₈₇(SO₄)₃电池在4.5 V下运行展现出延长的使用寿命,在1000 mA g⁻¹下循环16,500次后保持88.2%的容量(扣式电池),以及在100 mA g⁻¹下循环500次后保持93.9%的容量(软包电池)。该研究将为高能量密度电池等领域设计耐用电解液和界面相提供启发。
人工智能(AI)的迅猛发展和可再生能源的广泛开发加剧了对电网规模储能系统的需求。资源丰富的钠离子电池(SIBs)凭借克服锂资源稀缺和价格波动带来的可持续供应限制,已获得显著关注并取得了实质性的产业化进展。集成金属钠负极和高容量正极的先进钠离子电池有望提供与商用锂离子电池相当的能量密度并具有长循环耐久性。然而,从正极晶体中完全提取Na⁺需要高工作电压,这给电解液的相容性带来了严峻挑战。在深度脱钠过程中,从氧化还原对的eg轨道移走电子会引发严重的表面空穴累积和费米能级显著下移,产生强亲电性正极表面,从而引发对自由溶剂和Na⁺溶剂化络合物的亲电攻击。以溶剂为主的分解会生成有机自由基,通过亲电加成-缩合反应形成富含低聚物的界面相。这些多孔界面相无法抑制电子隧穿和阻挡溶剂渗透,从而加速了持续的电解液消耗和容量衰减。此外,常规碳酸酯在高电压下阳极稳定性有限,会释放气体(如O₂、CO₂),进而引发寄生反应和安全危害。
电解液工程有望解决高压正极/电解液界面不稳定的问题。当前的方法侧重于单方面调控。例如,通过高浓度或局部高浓度电解液进行体相溶剂化调控,以形成溶剂分离的接触离子对或离子聚集体;使用含氟添加剂(如氟代碳酸乙烯酯、氟苯)进行界面设计,以牺牲性预制界面相。尽管类似于局域化生物保护,但这些方法在实际应用中存在显著的权衡,包括离子电导率下降、粘度增加和负极不相容性。从根本上说,界面氧化遵循吸附介导的优先分解机制,其中溶剂经历竞争的静电力作用,如强烈的Helmholtz层电势与Born溶剂化能约束。这种动态的体相-界面耦合需要对体相溶剂化结构进行协同调控,以最小化高能构象,并通过阴离子优先的分解动力学实现定向界面组装。此外,阐明界面相组分的钝化行为,特别是它们与电压相关的电子/离子传输特性,对于全面的电池稳定性同样重要。
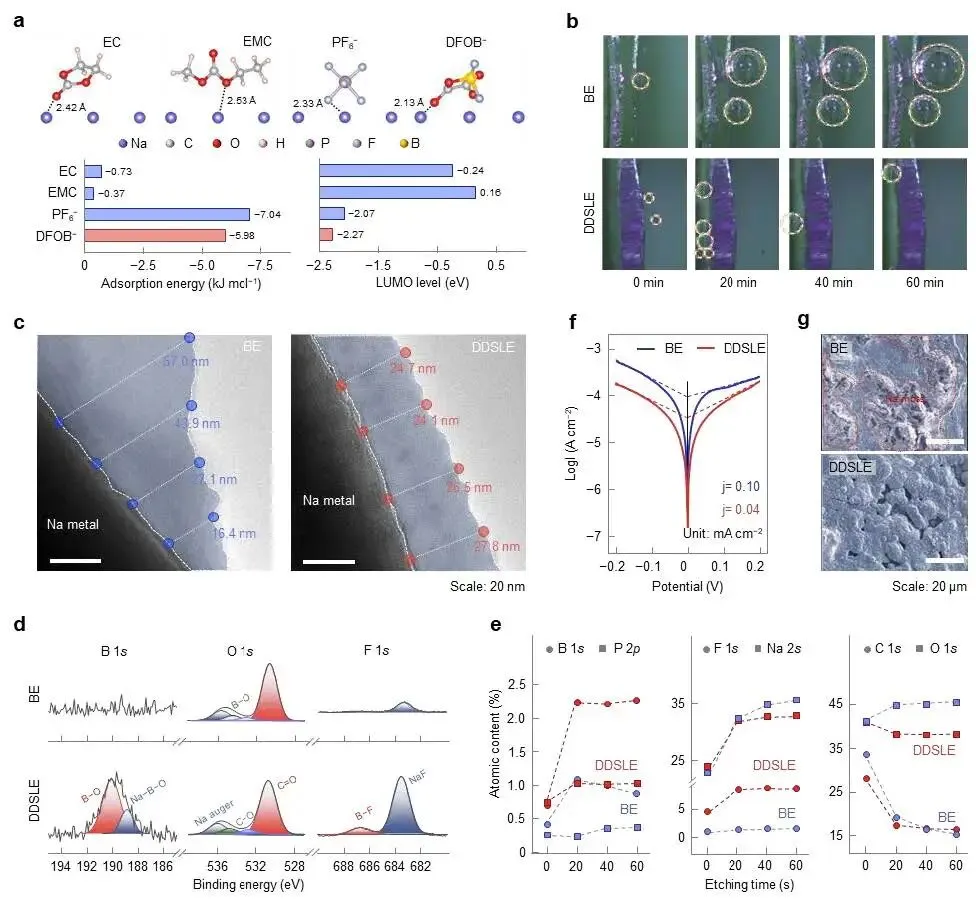
在此,该研究设计了一种溶剂锁定型电解液,以解决其在高电压下的电化学不稳定性。对称的二氟(草酸)硼酸根阴离子(DFOB⁻)作为工业电解液(1 M NaPF₆溶于碳酸乙烯酯(EC)/碳酸甲乙酯(EMC))中的添加剂,表现出与碳酸酯溶剂的弱配位作用,但对暴露Na⁺的正极表面有强结合亲和力。利用DFOB⁻阴离子的空间位阻和吸电子效应,所设计的电解液在体相电解液(域I)和正极界面(域II)的双域中实现了对溶剂分子的有效锁定。因此,研究人员将其命名为双域溶剂锁定电解液(DDSLE)。关键的是,DFOB⁻的优先分解生成了富含硼化物/氟化物的CEI组分,其具有宽禁带(>6.2 eV),提供了电子绝缘性和改善的抗氧化性。因此,DDSLE电解液在4.5 V级的Na||Na₂.₂₆Fe₁.₈₇(SO₄)₃扣式电池和软包电池中表现出良好的循环稳定性。
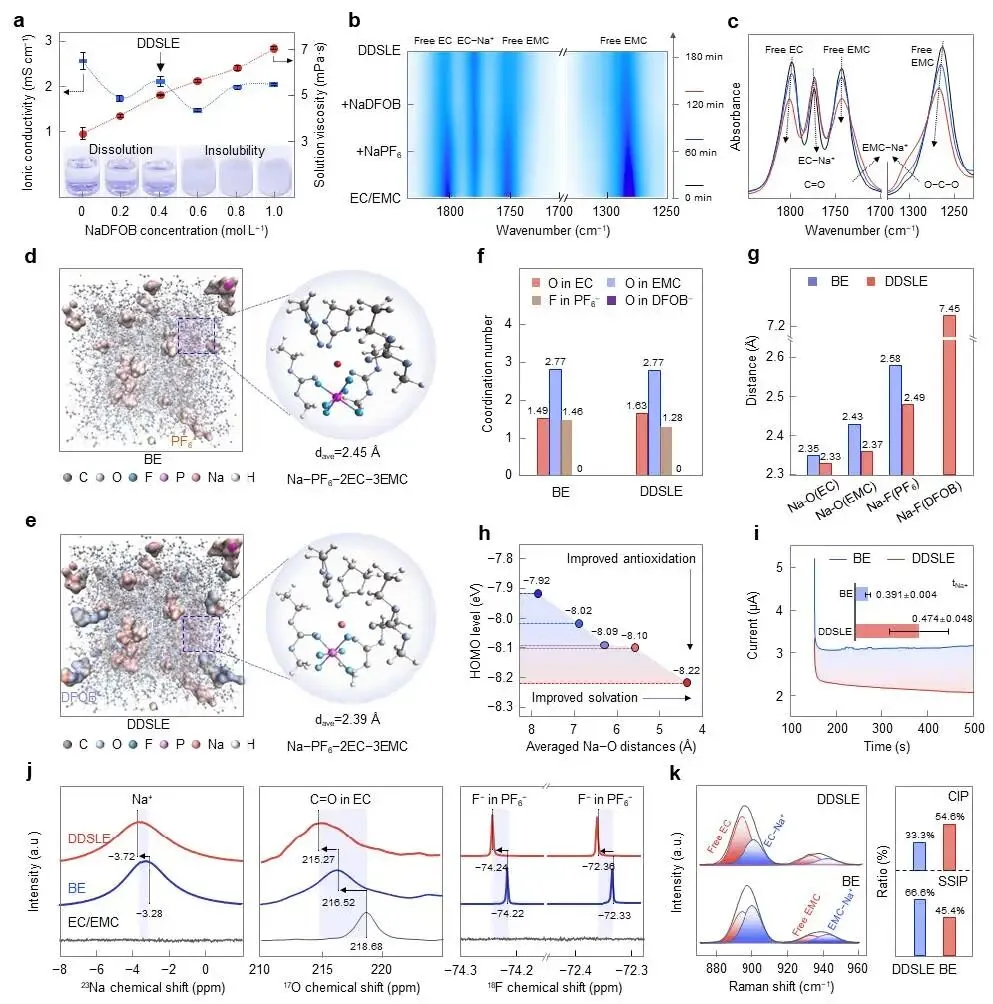
图1 | DDSLE电解液的化学性质与溶剂化结构。(a) 具有不同NaDFOB浓度的电解液的离子电导率、溶液粘度及数码照片。(b, c) 原位FTIR光谱追踪从BE到DDSLE电解液过程中盐在EC/EMC中的溶解过程。(d) BE和(e) DDSLE电解液的MD模拟快照,描绘了PF₆(黄色等值面)和DFOB(蓝色等值面);Na离子和EC/EMC分子以棒状表示。(f) 从MD模拟获得的平均配位数和(g) Na-O/F键长。(h) Na⁺溶剂化结构的HOMO能级随配位距离的变化。(i) Na||Na对称电池中BE和DDSLE电解液的计时电流曲线。插图:计算得到的Na⁺迁移数及其误差棒。(j) 纯溶剂、BE和DDSLE电解液的²³Na、¹⁷O和¹⁹F NMR谱。(k) BE和DDSLE电解液的拉曼光谱,及相应的溶剂化物种定量分析。误差棒代表拟合曲线的标准差,数据来自三次平行实验。
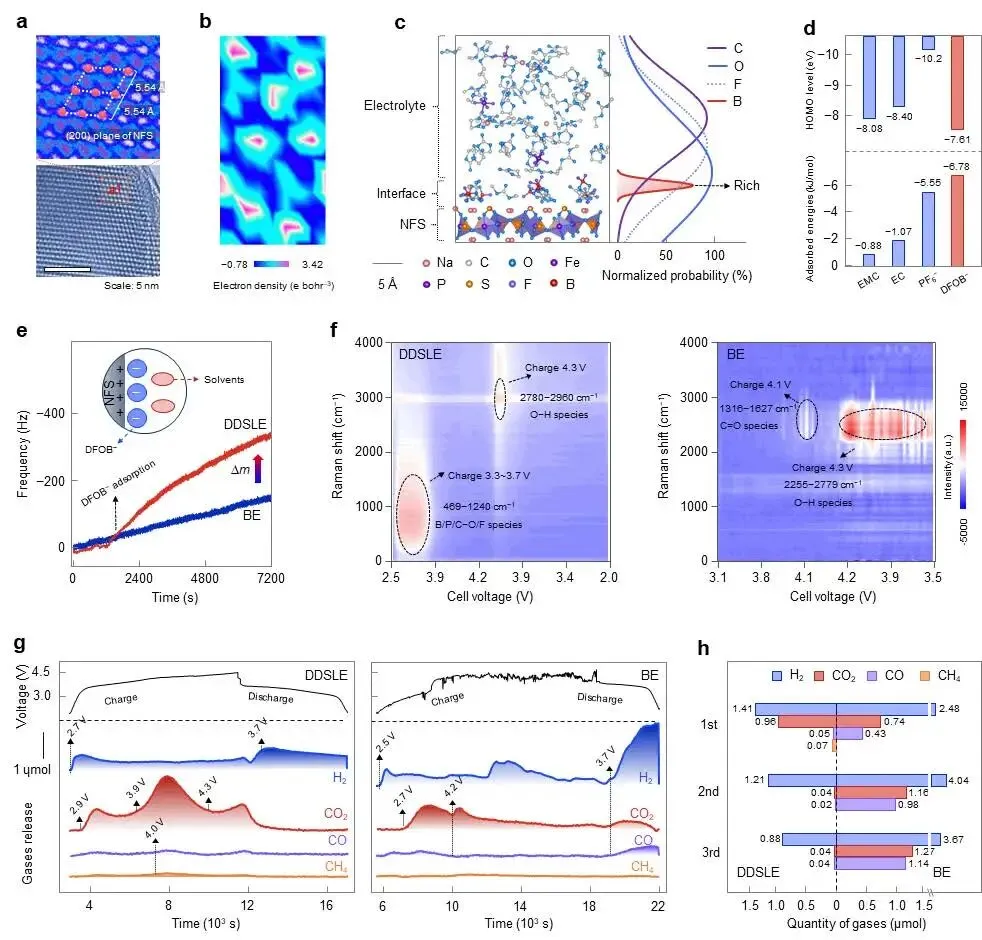
图2 | DDSLE电解液在正极界面的结构演化与反应。(a) NFS正极的HRTEM图像。(b) NFS(200)晶面的电子密度图。(c) NFS/DDSLE界面模型,以及阴离子和溶剂分子沿模拟盒子c轴的分布曲线。(d) 电解液各组分在NFS表面的计算吸附能及HOMO能级。(e) 通过EQCM测量的Na|NFS电池在50 mV s⁻¹和25 °C下的时间相关振荡频率偏移。(f) 采用DDSLE和BE电解液的Na|NFS电池在50 μA cm⁻²和25 °C下的原位拉曼光谱。(g) 采用DDSLE和BE电解液的Na|NFS电池在50 μA cm⁻²和25 °C下首次充电至4.5 V过程中的原位DEMS数据。(h) 计算得到的气体释放量。
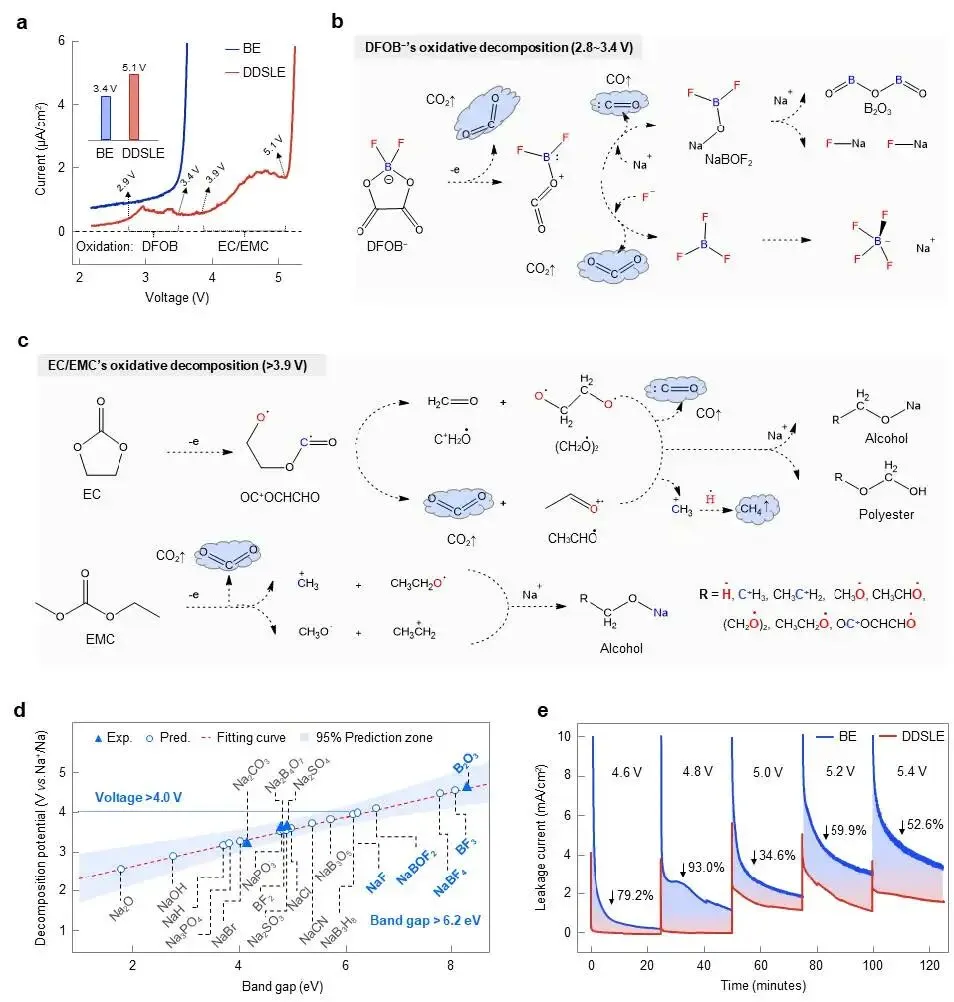
图3 | DDSLE电解液在NFS正极上被抑制的电化学反应活性。(a) Na|不锈钢片电池的LSV曲线。(b) DFOB⁻阴离子(2.8至3.4 V之间)和(c) EC/EMC溶剂分子(3.9 V以上)氧化分解过程中所提出的化学转化路径。(d) CEI中典型无机组分的分解电位及其对应的禁带宽度值。(e) 采用DDSLE和BE电解液的Na|NFS电池在高电压(>4.5 V)充电过程中的漏电流。
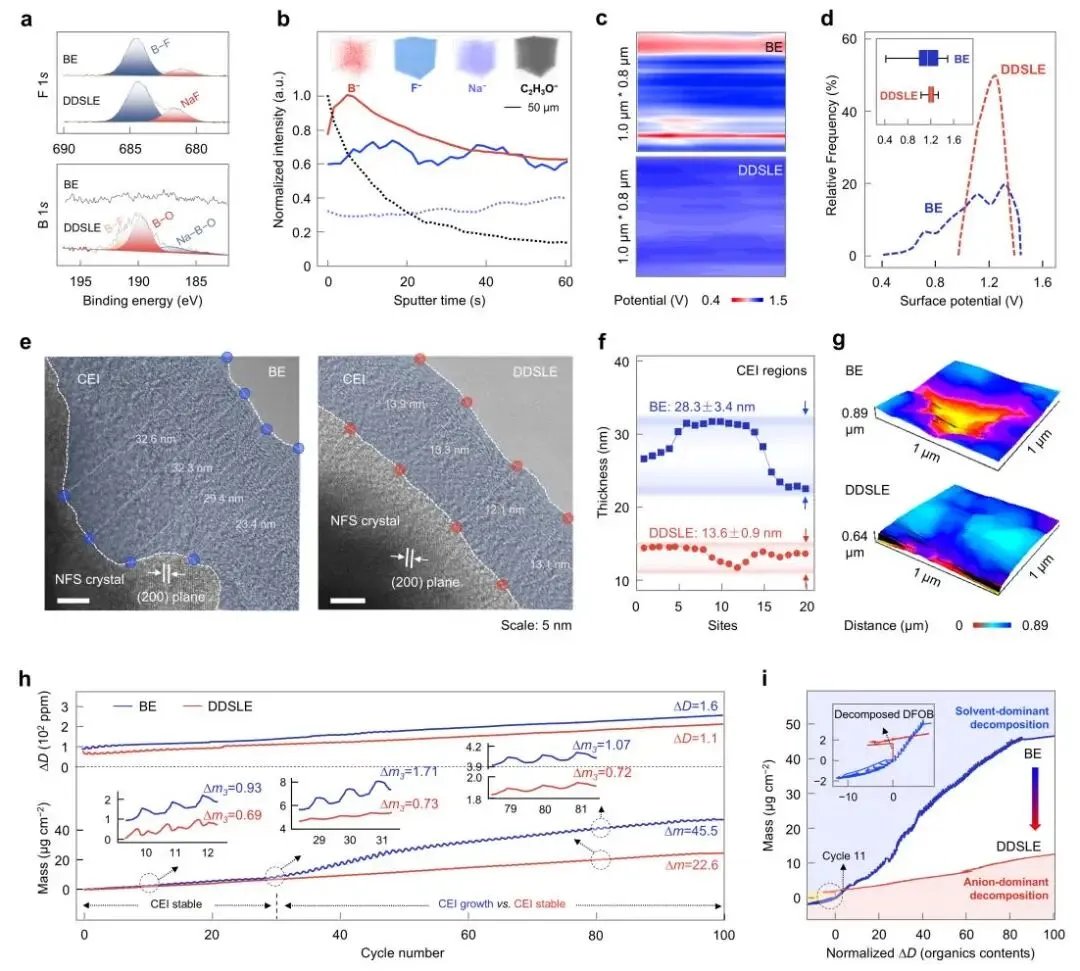
图4 | CEI的组成、结构与力学性能。(a) 循环后NFS正极的B 1s和F 1s XPS谱图。(b) DDSLE衍生的CEI中B⁻、F⁻、Na⁻和C₂H₃O⁻物种随溅射深度变化的TOF-SIMS 3D重构及相对强度。(c, d) 使用DDSLE和BE电解液循环后的NFS正极的静电势分布图。(e) 采用BE和DDSLE电解液形成的CEI的Cryo-TEM图像。(f) CEI的厚度分布。(g) 使用DDSLE和BE电解液循环后的NFS正极的表面粗糙度分布图。(h) 通过原位EQCM记录的Na|NFS电池在CV测量(50 mV s⁻¹,25 °C)中多个循环期间质量(Δm)和能量耗散(ΔD)随时间的变化。(i) 质量变化与能量耗散之间的相关性。图a-g中的电极是在Na|NFS扣式电池中以10 mA g⁻¹测试的。循环3次后,在2.0 V的放电状态下拆解电池。
图5 | 界面化学与SEI结构。(a) 吸附在钠金属表面的电解液组分的结构,以及它们的吸附能和LUMO能级。(b) 采用DDSLE和BE电解液的Na||Na对称电池在50 μA cm⁻²和25 °C下钠金属表面的原位光学显微镜图像。(c) 在BE和DDSLE电解液中形成的SEI的TEM图像。(d) 循环后钠负极的B 1s和F 1s XPS谱图。(e) 随XPS溅射深度变化的元素组成演变。(f) 采用DDSLE和BE电解液的Na||Na对称电池的Tafel曲线。(g) Na||NFS电池中循环500次后的钠金属表面的SEM图像。图c、d、e、g中的电极是在Na||NFS扣式电池中以10 mA g⁻¹测试的。循环3次后,在2.0 V的放电状态下拆解电池。
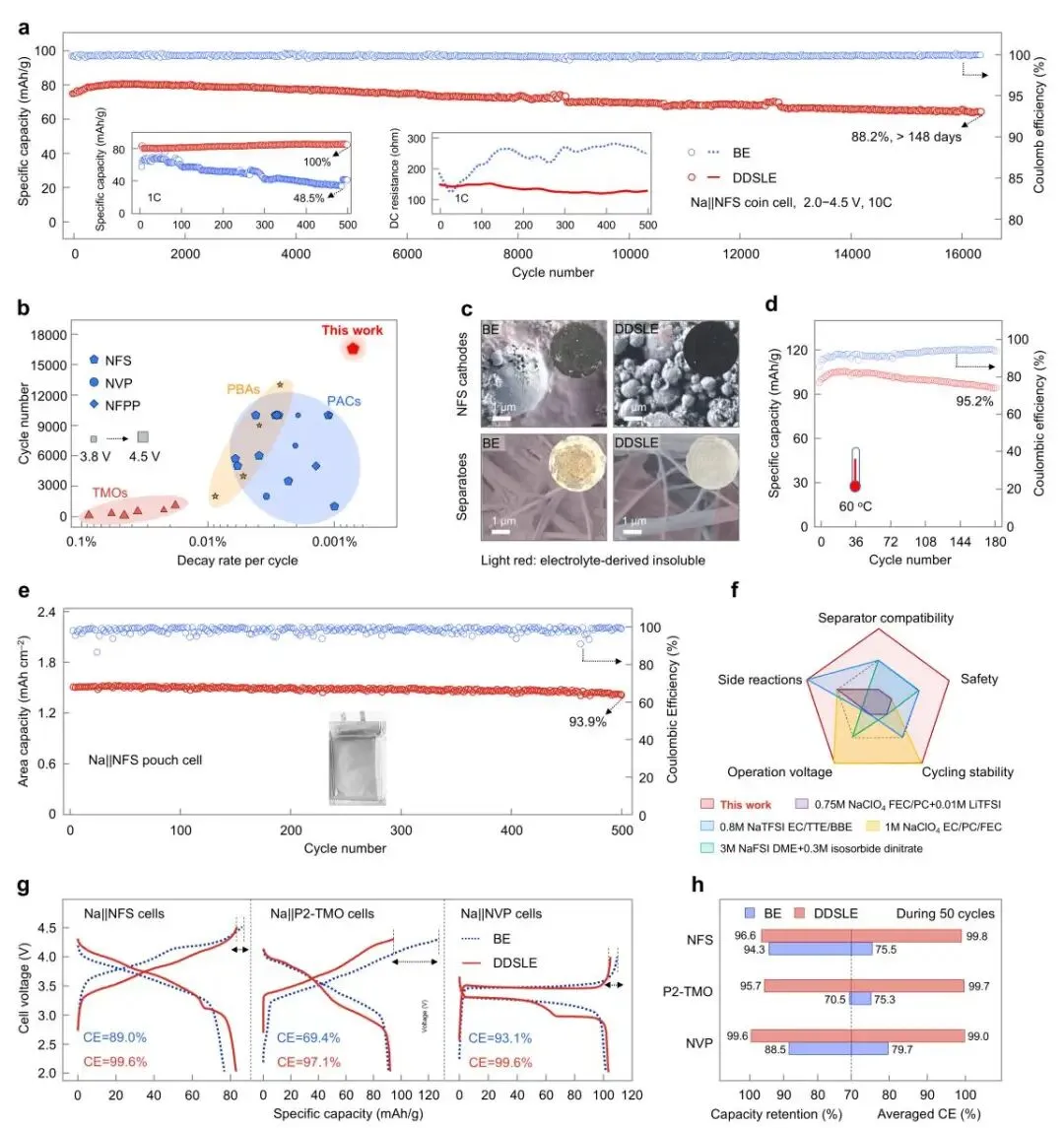
图6 | 采用DDSLE电解液的钠金属扣式电池和软包电池的电化学性能。(a) Na||NFS扣式电池在1000 mA g⁻¹和25 °C下的长期循环性能。插图:在1000 mA g⁻¹和25 °C下的循环性能及直流内阻。(b) Na||NFS电池与最先进的钠金属电池的性能对比。(c) Na||NFS扣式电池在1000 mA g⁻¹和25 °C下循环500次后的正极和隔膜的SEM图像。插图:拆解后相应电池的极片和隔膜的照片。该电极在Na||NFS扣式电池中以1000 mA g⁻¹进行测试,电池在循环500次后于2.0 V停止并拆解。(d) Na||NFS扣式电池在100 mA g⁻¹和60 °C下的循环性能。(e) Na||NFS软包电池在100 mA g⁻¹和25 °C下的循环性能。(f) 比较钠离子电池系统中代表性电解液性能的雷达图。本图所示文献数据的来源见补充表4。(g) 采用DDSLE和BE电解液的Na||NVP、Na||P2-TMO和Na||NFS扣式电池在100 mA g⁻¹和25 °C下的恒流充放电曲线。(h) 电池在初始50次循环中的容量保持率和平均库仑效率统计。
总之,该研究提供了一种双域溶剂锁定电解液(DDSLE),以解决工业NaPF₆/碳酸酯基电解液在高电压(>4.3V)下的长期不稳定性挑战。通过引入对称二氟(草酸)硼酸根阴离子(DFOB⁻),在体相溶液内构建了溶剂增强的Na⁺溶剂化结构(域I),并在正极界面构建了阴离子富集的屏蔽层(域II),从而有效限制了溶剂分子的自由度和反应活性。因此,研究人员将其称之为双域溶剂锁定电解液。这种溶剂锁定构型将电解液分解路径从溶剂主导演变为阴离子主导,促进了稳定的界面相形成。在正极侧,具有富含硼化物/氟化物内层和富含聚酯/醇盐外层的集成CEI有效抑制了电子隧穿,将正极接触电位降低了16.7%,漏电流降低了34.6−79.2%。实验与理论分析相结合,建立并验证了分解电压与CEI组分禁带宽度之间的线性相关性,确定了电子隧穿电阻是高压界面稳定性的主要决定因素。关键的是,禁带宽度超过6.2eV的硼化物/氟化物被证明能有效抑制4.0V以上的电子隧穿。同时,在钠负极上形成了薄而均匀的SEI,实现了可逆的钠沉积。这些协同效应显著增强了电解液在高电压下的稳定性,表现为阳极稳定极限的提高(从3.4V到5.1V)、库仑效率的提升以及过充和气体释放的减少(减少46.6%)。该DDSLE电解液展示了与钠负极以及多种正极(包括Na₂.₂₆Fe₁.₈₇(SO₄)₃、Na₃V₂(PO₄)₃和Na₀.₇₂Ni₀.₃₂Mn₀.₆₈O₂)的相容性。组装的Na||Na₂.₂₆Fe₁.₈₇(SO₄)₃电池在4.5V的截止电压下提供了长循环稳定性,在扣式电池中循环16,500次后实现88.2%的容量保持率,在软包电池中循环500次后实现93.9%的容量保持率。该研究为下一代高电压电池的系统性电解液工程开辟了一条颇具前景的途径。


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
