华东理工大学朱明辉/郑州大学李迪迪ACS Catal.:原位光谱表征技术揭示不同钴负载量的铜基催化剂在甲醇重整制氢中的构效关系
- 2026-05-09 04:59:01

■ 围绕科研服务科研
特别说明:
感谢您的关注,因文章由英文论文翻译过来,如存在误差,请您致电联系小编,小编将第一时间修正。
联系电话:19392795346(微信同)
技术支持:15311208010(微信同)

1
文章摘要
探索铜与金属氧化物之间的金属-载体相互作用对于设计铜基催化剂至关重要。本研究发现,在还原条件下可以连续调节金属铜与钴氧化物之间的相互作用。对不同钴负载量的Cu/Al₂O₃催化剂进行一系列原位表征。结果表明,钴物种的化学状态和空间分布随钴含量变化而发生明显变化。钴负载量较低时,略微增强了铜的分散性并有限的调节电子。随着钴负载量增加,可还原的CoOx物种迁移并部分包裹铜纳米颗粒,形成强金属-载体相互作用结构并产生大量Cu-CoOx界面位点,从而增强了甲醇蒸汽重整制氢的反应活性。而掺杂过量的钴则导致CoOx还原为金属钴,使甲醇加速裂解并降低了甲醇水蒸汽重整制氢的反应活性和CO₂选择性。在本研究中,Cu20CoAl催化剂具有最佳性能,在相同的铜负载量下,其H₂产率是CuAl的1.5倍。
2
背景介绍
铜在低温下具有高活性和低一氧化碳选择性,被广泛用作甲醇水蒸汽重整制氢反应的催化剂,氧化铝作催化剂载体。除载体外,通常还会向催化剂中添加助剂。利用金属纳米颗粒与金属氧化物助剂之间的相互作用可以改变催化剂的表面微观结构并促进形成新位点,从而提高催化剂的活性、选择性和稳定性。强金属-载体相互作用是指可还原的氧化物载体或助剂在还原气氛下迁移到金属表面,形成气体可渗透的覆盖层,从而改变催化剂表面结构和活性位点分布。研究表明,强金属-载体相互作用也存在于各种铜基催化剂体系中。深入研究铜与强金属氧化物之间的金属-载体相互作用是催化领域的重点。探索具有强金属-载体相互作用的新型Cu-CoOₓ体系对于合理设计此类工业催化剂至关重要。钴作为助剂能有效提高铜的分散性并改善催化剂的还原性。本研究探讨了不同钴负载量对铜基催化剂在甲醇蒸汽重整反应中结构、界面化学和催化性能的影响。制备了一系列铜负载量固定、钴含量不同的CuO-xCo₃O₄-Al₂O₃催化剂,并利用一系列原位表征技术深入讨论其结构-活性关系。
3
图文解析
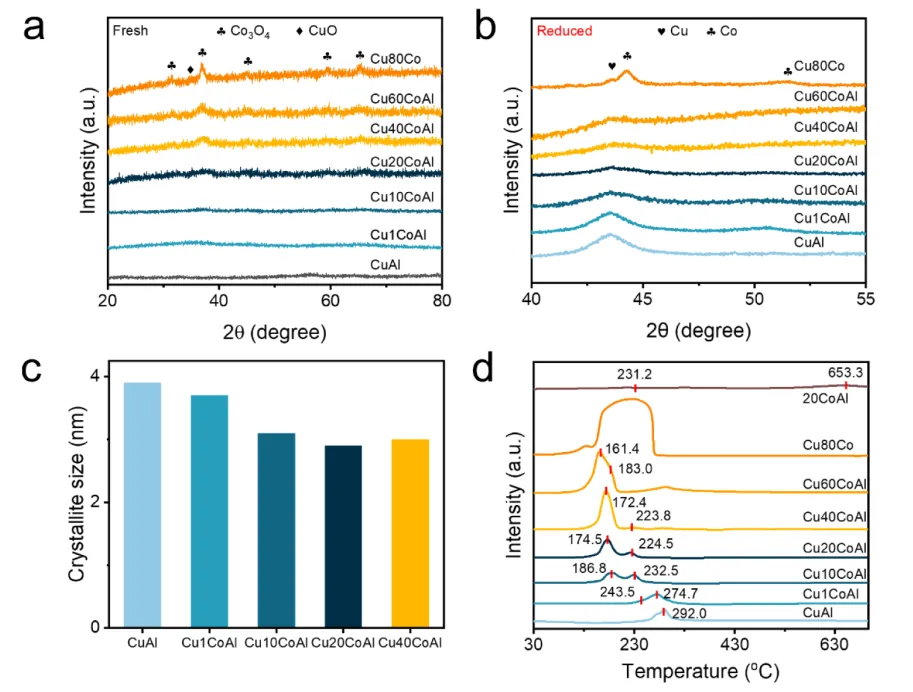
图1.CuxCoAl催化剂的晶体结构和形貌。(a) 未还原和(b) 还原的CuxCoAl催化剂的原位XRD图谱。(c) 通过原位XRD获得的Cu微晶尺寸。(d) 未还原的CuₓCoAl催化剂的H2-TPR分布图。
采用原位XRD表征CuxCoAl催化剂的结构(图1a)。未还原的催化剂中出现Co₃O₄衍射峰。增加Co负载量后,Co₃O₄的衍射峰强度逐渐增强。在35.5°处出现CuO衍射峰,强度相对较弱,表明CuO物种分散良好。在未还原的催化剂中未出现Al₂O₃的特征峰,表明Al₂O₃以无定形形式存在。催化剂在300°C下还原1h后,Co₃O₄和CuO的特征峰完全消失,同时出现金属Cu的衍射峰(图1b)。在低Co负载量和中等Co负载量的催化剂(x=1-60)中未检测到金属Co或其他钴氧化物的衍射峰,表明Co物种可能以高度分散或非晶形式存在。在Co含量较高的(Cu80Co)样品中,出现金属Co的衍射峰,说明Co被还原为金属态。
根据XRD数据计算CuxCoAl催化剂的平均Cu晶粒尺寸(图1c)。研究表明,掺杂适量的Co(如Cu20CoAl)将减小Cu晶粒尺寸,提高Cu的分散性;掺杂过量的Co(Cu80Co)将导致Cu颗粒尺寸增大。这种差异表明,除晶粒尺寸外,其他因素在决定Cu分散度方面起关键作用。具体而言,部分可还原的CoO物种在还原过程中可能迁移到金属Cu表面,形成覆盖层。这个覆盖层减少了暴露的Cu活性位点的数量。图1d展示了不同Co含量催化剂的H2−TPR还原曲线。随着Co含量增加,CuO的还原峰逐渐向低温移动,引入Co促进了CuO还原,形成Cu–Co协同还原的界面结构,增强了还原性。
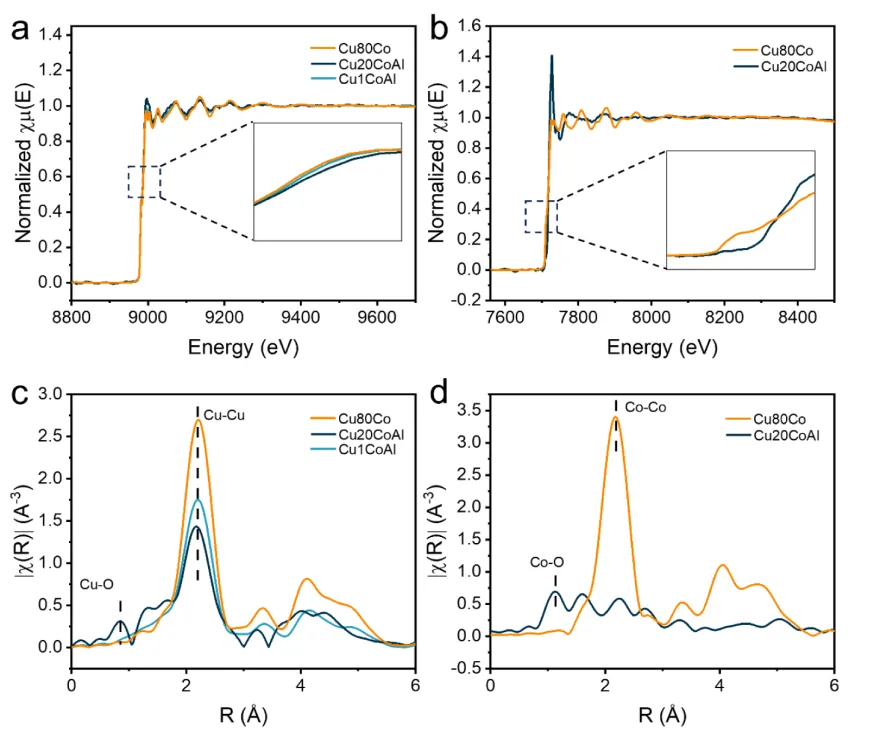
图2.CuxCoAl催化剂的分子结构。(a) 还原后的CuxCoAl催化剂的Cu K边X射线吸收近边结构(XANES)。(b) 还原后的CuxCoAl催化剂的Co K边XANES。(c) 还原后的CuxCoAl催化剂的Cu K边扩展X射线吸收精细结构(EXAFS)光谱。(d) 还原后的CuxCoAl催化剂的Co K边EXAFS光谱。
采用原位X射线吸收谱研究还原后的Cu1CoAl、Cu20CoAl和Cu80Co催化剂的局部结构演变(图2a、b)。Cu K边XANES结果表明,还原后的Cu20CoAl催化剂的吸收边相对于其他样品向高能量方向移动,表明其Cu物种的整体氧化态更高。Co K边XANES光谱揭示了不同催化剂中Co物种的差异:Cu1CoAl样品中几乎无法检测到Co信号。与Cu20CoAl相比,Cu80Co催化剂的吸收边向低能量方向移动,表明Co以较低的氧化态存在,即含有更高比例的金属Co物种。
在Cu K边EXAFS光谱中(图2c),Cu20CoAl的Cu-O配位峰较强,Cu-Cu配位峰相对较弱。这表明Cu原子对O原子具有强亲和力,并且存在与CoOx的强界面相互作用。相比之下,Cu1CoAl和Cu80Co催化剂主要存在Cu-Cu配位峰,Cu80Co的Cu-Cu峰强度高于Cu1CoAl。这些结果表明,当Co负载量超过最佳值时,会导致Cu团聚以及Cu-CoOₓ界面相互作用失效。在Co K边光谱中(图2d),Cu1CoAl的信号忽略不计,Cu20CoAl催化剂中存在Co-O配位峰,表明Co物种主要以氧化态存在。同时,Cu80Co催化剂表现出一个强的Co-Co配位峰,对应于Co物种的深度还原和团聚。
测试结果分析
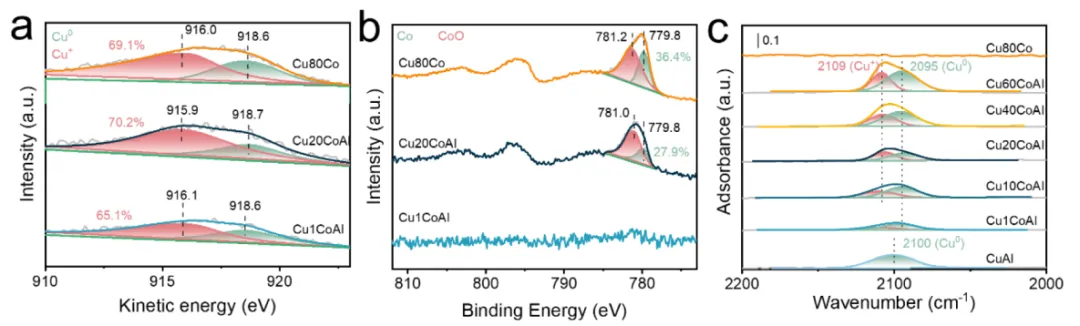
图3.(a) 通过准原位XPS获得的还原CuxCoAl催化剂的Cu LMM俄歇谱和(b) Co 2p XPS区域。(c) 还原后的CuxCoAl催化剂的原位CO吸附-红外光谱。
通过准原位X射线光电子能谱和原位CO吸附-红外光谱分析还原后的催化剂表面化学状态和活性位点。由于XPS的Cu 2p峰无法有效区分Cu0和Cu+。使用Cu LMM俄歇光谱来区分Cu0(918.6 eV)和Cu+(916.0 eV)(图3a)。通过拟合分峰计算表面Cu+物种的比例。结果表明,Cu20CoAl的Cu+比例最高,这归因于其最显著的金属-载体相互作用以及由此产生的大量高活性Cu-CoOx界面位点。从Co 2p光谱中解析CoO(778.2-785.0 eV)和金属Co(778.6-782.8 eV)的相对含量(图3b)。对于Cu1CoAl催化剂,由于其Co含量低,Co 2p峰强度极弱。与Cu20CoAl(27.9%)相比,Cu80Co催化剂表面表现出更高比例的Co0(36.4%)。
如图3c所示,使用CO作为探针分子,在室温下进行原位漫反射红外傅里叶变换光谱表征,探测催化剂表面的Cu0和Cu+位点。还原后的CuAl催化剂仅在2100 cm-1处检测到单一峰,这归因于CO在Cu0位点上吸附,表明Cu与Al2O3载体之间的界面对于CO吸附而言较弱。还原后的CuxCoAl催化剂在2095 cm-1和2109 cm-1处出现两个谱峰,分别归属于CO在金属Cu0位点和Cu+位点上的吸附。Cu0-CO峰红移表明引入Co调节了Cu的电子结构,增强了反馈键。Cu+-CO峰的出现直接证明存在Cu−CoOx界面位点。
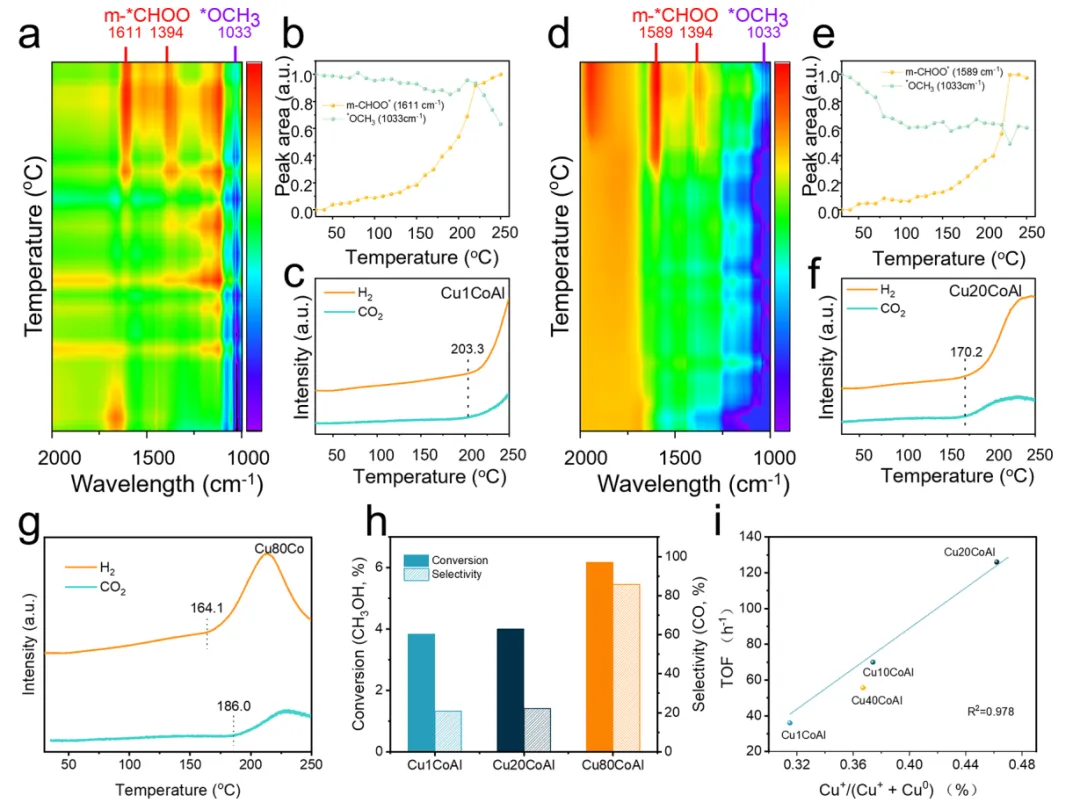
图4.(a) 对于还原后的Cu1CoAl,以5 °C/min的升温速率将温度升至250℃期间的原位漫反射红外傅里叶变换光谱。(b) 还原后的Cu1CoAl表面甲氧基(1033 cm-1)和表面甲酸根(1611 cm-1)的积分峰面积随温度的变化。(c) Cu1CoAl在升温过程中CO2和H2的质谱信号。(d) 对于还原后的Cu20CoAl,以5 °C/min的升温速率将温度升至250℃期间的原位漫反射红外傅里叶变换光谱。(e) 还原后的Cu1CoAl表面甲氧基(1033 cm-1)和表面甲酸根(1589 cm-1)的积分峰面积随温度的变化。(f) Cu20CoAl在升温过程中CO2和H2的质谱信号。(g) Cu80Co在升温过程中CO2和H2的质谱信号。(h) 还原后的CuxCoAl催化剂用于甲醇分解反应的CH3OH转化率和CO选择性。(i) 原位CO-DRIFTS测定的Cu+物种在Cu-CoOx界面处的分数与TOF值之间的相关性,以及相应的相关系数。
甲醇蒸汽重整反应的反应机理已得到广泛研究,最常提出的途径是甲醇分解途径和甲酸盐途径。为确定在CuxCoAl催化剂上的主要反应途径,进行原位漫反射红外傅里叶变换光谱-质谱联用实验(图4a-g)。在升温过程中,实时监测催化剂表面的吸附中间体(红外信号)和生成的气相产物(质谱信号),从而推断反应路径。Cu1CoAl和Cu20CoAl催化剂在低温下表面存在中间体,如2923,2816和1033 cm-1处的甲氧基(*OCH3)。随着温度升高,甲氧基物种逐渐消失,同时逐渐形成单齿甲酸盐(*CHOO)。这些原位观察表明,CuxCoAl催化剂上的甲醇蒸汽重整反应涉及甲氧基物种转化为甲酸盐中间体,随后甲酸盐分解为CO2和H2。
热重-质谱联用分析表明,不同催化剂的产物演变存在明显差异。对于Cu1CoAl,在203.3℃出现H2,CO2信号。对于Cu20CoAl,在170.2℃同时出现两种产物,这表明甲醇和水在其表面更容易被活化。对于Cu80Co,在161.4℃下出现H2信号,在186.0℃出现CO2信号。这种产物的顺序演变表明反应机理发生转变。众所周知,甲醇通过脱氢途径分解会导致早期形成H2和*CO中间体,而甲酸盐途径涉及表面*CHOO物种分解同时生成H2和CO2。对三种代表性催化剂进行甲醇分解测试,进一步验证所提出的机理。如图4h所示,甲醇转化率的顺序为:Cu80Co(6.2%)>Cu20CoAl(4.0%)>Cu1CoAl(3.8%)。相比之下,CO选择性顺序为:Cu80Co(85.7%)>Cu20CoAl(22.1%)>Cu1CoAl(20.9%)。这些结果进一步证实,还原后的Cu80Co催化剂中生成的金属Co物种是促进甲醇直接分解为CO的关键。基于这些观察结果,可以推断Cu1CoAl和Cu20CoAl上的甲醇蒸汽重整反应主要遵循甲酸盐反应途径。然而,在Cu80Co催化剂上,除了甲酸盐途径外,还有金属Co催化的甲醇分解机理,后者导致CO2选择性降低。
为明确Cu-CoOx界面位点的作用,建立了转化频率值与通过CO-漫反射红外傅里叶变换光谱测定的Cu+物种比例之间的定量相关性(图4i)。转化频率值与(Cu+-CO)和(Cu0-CO)峰的强度比呈现正相关。这表明Cu+物种的形成主要与Cu-CoOx界面位点相关,在提高本征催化性能方面起关键作用。Cu20CoAl催化剂表现出最高比例的Cu+物种和相应的最高转化频率值,证明了适度的Co负载量通过强金属-载体相互作用效应形成了丰富且高活性的Cu-CoOx界面。这表明Cu-CoOx界面位点是甲醇水蒸气重整反应的本征活性中心。界面位点越多,催化剂的本征活性越高。
原位实验细节
In situXRD实验表征细节
采用原位X射线衍射技术表征催化剂的晶相演变和晶粒尺寸变化。使用布鲁克D8型X射线粉末衍射仪,其配备有Cu Kα射线源(波长λ=0.154 nm),操作电压和电流分别为40 kV和40 mA。实验中,将催化剂粉末置于原位反应池中,在300°C、10% H₂/Ar气氛下进行原位还原1 h。XRD图谱的采集范围为2θ=30°至80°,扫描步长为0.02°。
In situ XAS实验表征细节
在上海同步辐射装置(SSRF)的BL14W1光束线以透射模式进行Cu K边和Co K边的原位X射线吸收光谱表征。使用固定出口双晶Si(111)单色仪收集扩展X射线吸收精细结构(EXAFS)数据。采集数据之前,将样品压制成自支撑片并放置在原位反应池中,然后将样品在10% H2/Ar混合物中在300℃下原位还原1 h。使用Athena和Artemis软件包进行数据处理和分析。
Quasi in situ XPS实验表征细节
在配备单色Al kα辐射源(1486.6 eV)的Thermo ESCALAB 250Xi光谱仪上进行准原位X射线光电子能谱分析。催化剂在连接的预处理室中进行还原。还原后,将预处理室抽真空,然后将样品直接转移到真空分析室中,整个过程避免暴露在空气中。所有光谱均以284.8ev处的C 1s峰为参考进行结合能校准。
In situ CO−DRIFTS 实验表征细节
在配备漫原位反射池的PerkinElmer Frontier光谱仪上进行原位CO吸附漫反射红外傅里叶变换光谱表征。催化剂在300℃、10% H2/Ar气氛中原位还原30 min。随后,将样品在Ar气氛中冷却至室温。最后,将30 mL/min的0.05% CO/He引入池中30 min。以4 cm-1的分辨率采集光谱,每个光谱代表64次扫描的平均值。
4
结 论
本研究阐明了不同钴负载量的CuxCoAl催化剂在甲醇水蒸汽重整反应中的构效关系。一系列原位表征证实,钴负载量是决定催化剂结构演变和界面化学的关键因素。在低钴含量下,钴的掺入仅略微改善了铜的分散度,对活性的影响可忽略不计。随着钴负载量的增加,可还原的CoOx物种迁移并部分包裹Cu纳米颗粒,产生强金属-载体相互作用效应,并形成丰富的Cu-CoOx界面位点。这些界面作为高活性中心,通过甲酸盐途径促进甲醇和水的活化。这导致催化活性呈火山形趋势,其中Cu20CoAl被确定为最佳组成比例。过量的钴负载量导致Cu-CoOx固溶体深度还原,形成金属钴纳米颗粒。这些钴纳米颗粒活性物种优先催化甲醇裂解反应,从而显著降低甲醇蒸汽重整反应的CO2选择性和活性。这些结果突出了Cu-CoOx界面在控制甲醇蒸汽重整反应性能中的关键作用,并表明合理调节钴含量是平衡Cu-CoOx界面位点的形成与钴深度还原为金属态的关键策略。
原文链接:
Jiang Z, Gu H, Yan H, et al. Tunable Metal–Support Interactions between Copper and Cobalt Oxides for Enhanced Methanol Steam Reforming[J]. ACS Catalysis, 2026.
DOI:10.1021/acscatal.5c08720
https://doi.org/10.1021/acscatal.5c08720
原位测试中心可承接各种原位测试业务
如需转载或合作
请联系我们
联系方式:19392795346(微信同)
技术支持:15311208010(微信同)
联系邮箱:Yuanweiren2022@126.com
原位测试服务



微信公众号 官方微信号
推荐阅读

