郑州大学卢思宇/常江伟Angew:定制化O-O自由基耦合路径助力高密度Fe-N-C催化剂实现稳定工业规模水氧化!
Customized O-O Radical Coupling Route on High-Density Fe-N-C Catalysts for Stable Industrial-Scale Water Oxidation | Angewandte Chemie International Edition, 2026 |
| |
| 高密度 Fe-N-C 单原子催化剂;碱性析氧反应;AEMWE |
| 通过提升 Fe 位点密度调控 *OH 覆盖度,使 OER 从 AEM 转向 OPM,并获得工业级电解稳定性。 |
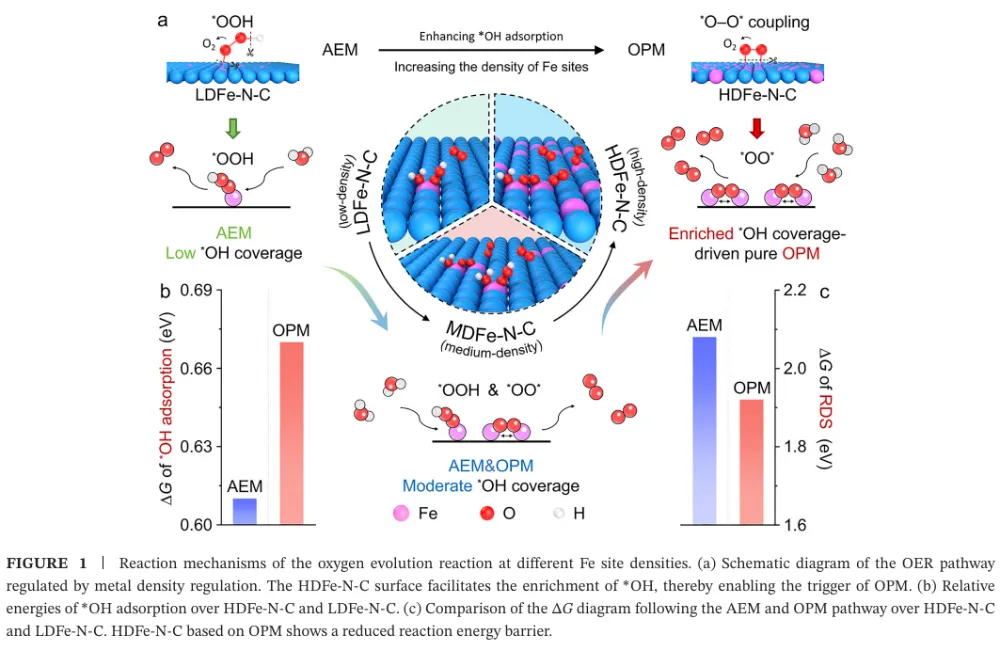
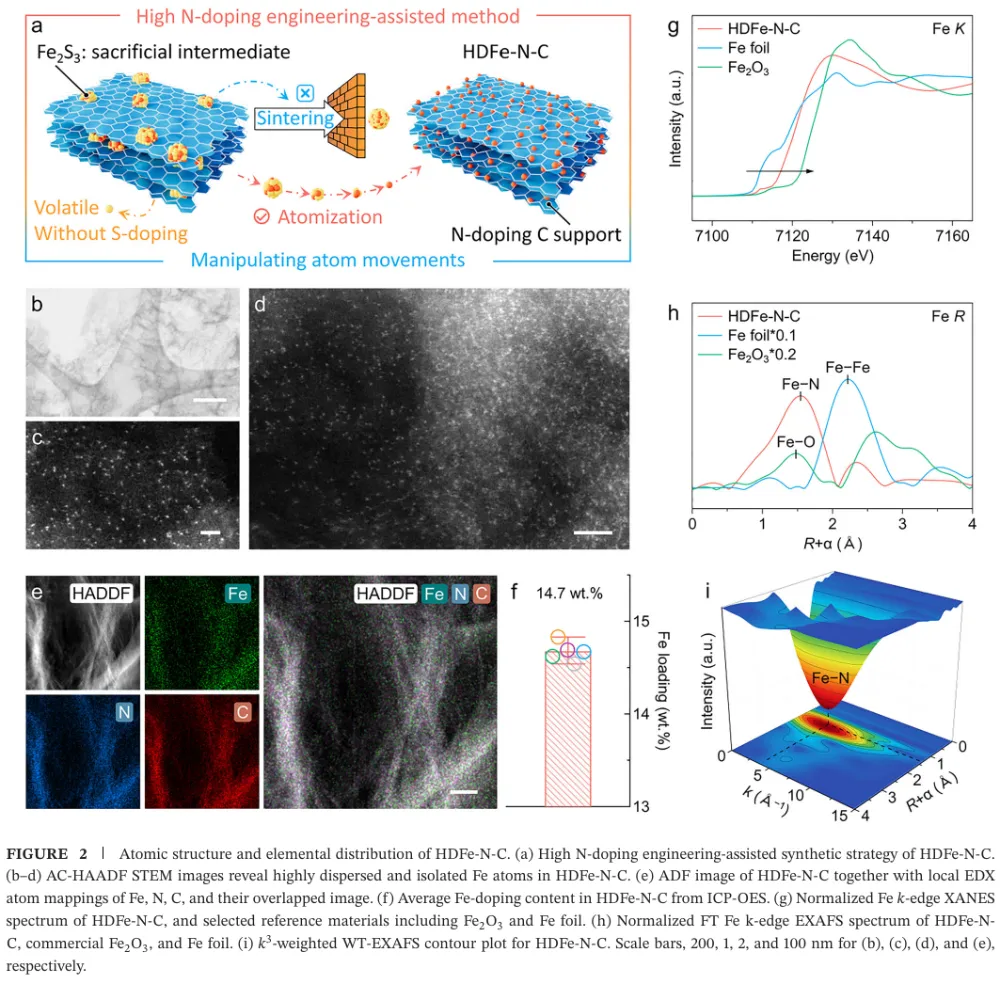
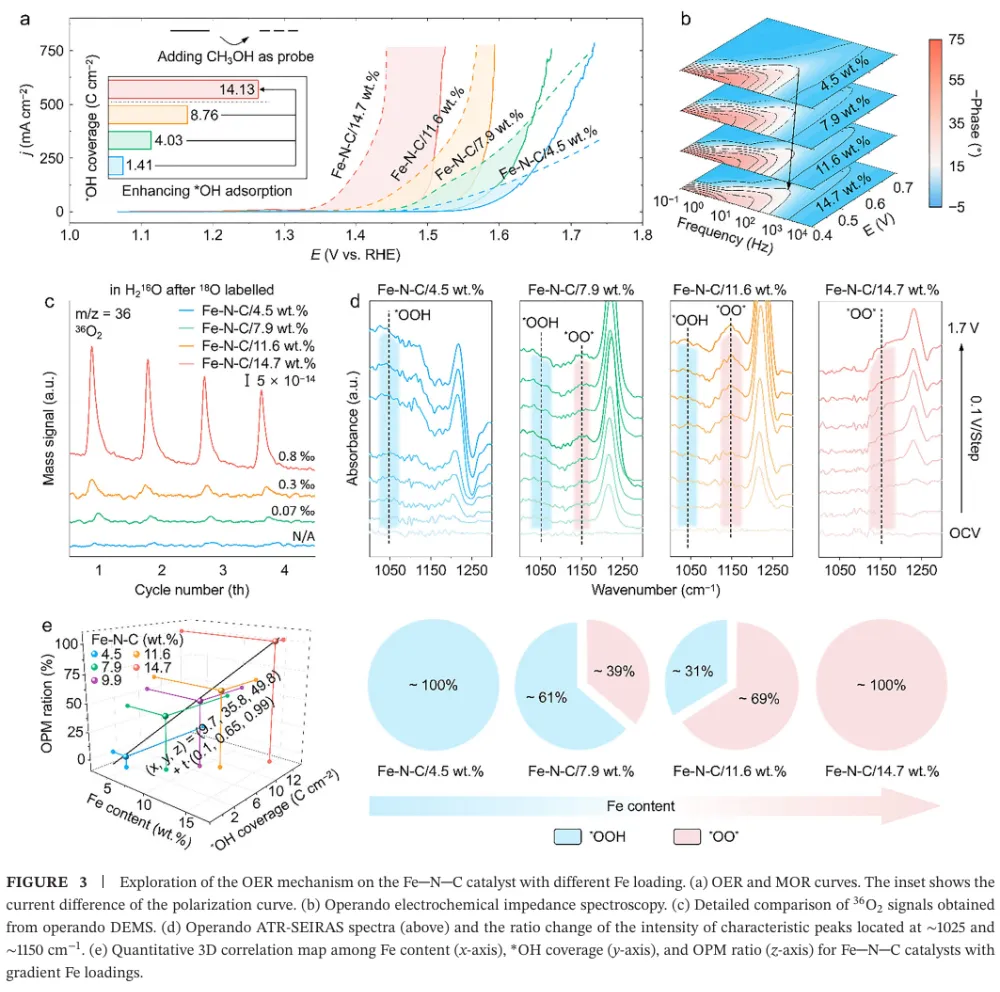
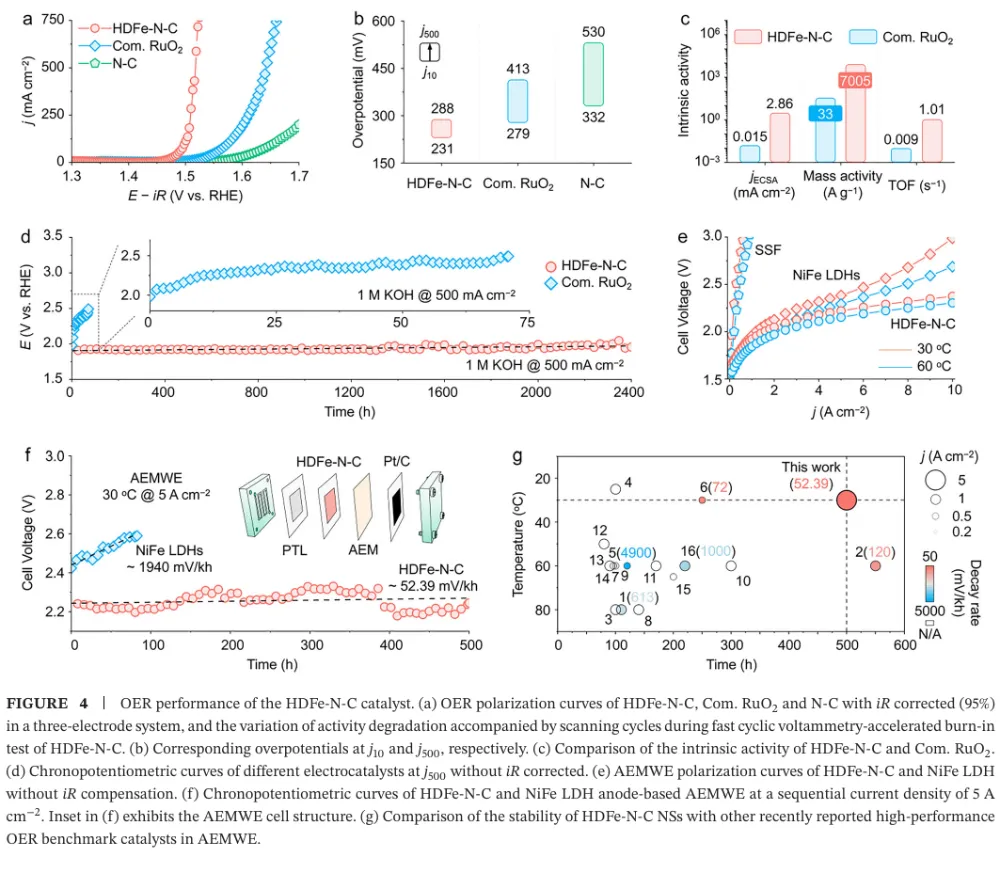
大规模绿氢需要低成本、耐久的 AEMWE 阳极 OER 催化剂 → 工业电流密度下 OER 动力学慢、催化层传质阻力与长期稳定性不足成为瓶颈 → 传统 M-N-C 单原子催化剂虽有高原子利用率,但金属负载通常偏低,活性位点相互孤立,往往被限制在吸附演化机制 AEM 的线性标度关系中 → 本文提出通过提高 Fe-N-C 中 Fe 单原子位点密度,增强表面 *OH 覆盖并缩短相邻活性位距离 → 高密度 Fe 位点促使相邻 *O 物种直接发生 *O-*O 自由基偶联,推动 OER 路径从 AEM 转向氧化物路径机制 OPM → DFT、MOR、电化学阻抗、同位素 DEMS、ATR-SEIRAS 与 XAS 共同验证机制转换 → HDFe-N-C 在 500 mA cm-2 下以 288 mV 过电位稳定运行 2000 h,并在 AEMWE 中实现 5 A cm-2、2.15 V 与 500 h 稳定运行。这篇文章真正有价值的地方,不只是报道了一个高负载 Fe-N-C 单原子催化剂,而是把“金属位点密度”从一个合成指标提升为反应路径调控变量。传统单原子催化剂常被理解为孤立位点体系,其优势在于位点明确、原子利用率高;但在工业级 OER 中,孤立位点同时带来低负载、厚催化层、位点协同不足和 AEM 线性标度限制等问题。本文抓住的核心矛盾正是:单原子催化剂如果一直停留在低密度孤立位点范式中,很难支撑安培级电解。作者的思路是把 Fe 位点做得足够密集,同时仍保持原子级分散,利用相邻 Fe-N4 位点之间的几何邻近和电子协同,创造 *O-*O 偶联条件。这使 OER 不再主要依赖孤立 Fe 位点上 *OH、*O、*OOH 的逐步吸附转化,而是转向相邻含氧中间体之间的直接偶联。这个机制转向解释了为什么高 Fe 负载不只是增加位点数量,还会改变单位位点的反应本质。文章的证据链比较完整:先用理论计算提出高密度 Fe 位点增强 *OH 吸附并降低 OPM 速控步能垒,再用结构表征证明 14.7 wt.% Fe 仍以 Fe-N4 单原子形式存在,随后构建 4.5、7.9、11.6、14.7 wt.% 的 Fe 负载梯度,通过原位/操作态谱学和同位素信号把 Fe 含量、*OH 覆盖度和 OPM 占比关联起来。最后再把材料放进高电流 OER 和 AEMWE 器件中验证可用性。需要注意的是,本文最强的说服力来自“机制证据与器件稳定性相互闭合”:OPM 被认为能避免 LOM 中晶格氧参与导致的结构流失,而作者确实展示了长时间 OER、AEMWE、启停循环和复杂工况下的稳定性。这种从机制到应用的闭环,使文章不只是一个性能排名工作,而是一个关于单原子催化剂如何突破工业化场景限制的设计范式。变量设计:作者以 Fe-N-C/4.5 wt.%、7.9 wt.%、11.6 wt.%、14.7 wt.% 构建金属负载梯度,把 Fe 位点密度作为唯一主线变量来追踪 OER 机制和性能变化。这样的设计避免了只比较最终材料而无法解释机制来源的问题。结构确认:HDFe-N-C 的 Fe 含量达到 14.7 wt.%,但 AC-HAADF-STEM、EDS、XANES、EXAFS 和 WT-EXAFS 均指向高度分散的 Fe-N4 构型,没有明显 Fe-Fe 或 Fe-O-Fe 聚集信号。高负载与单原子分散同时成立,是后续机制讨论的前提。机制证据:MOR 证明高 Fe 负载强化 *OH 吸附;operando EIS 显示与 *OH 吸附相关的界面弛豫增强;18O 标记 DEMS 中 36O2 随 Fe 负载升高而增强,说明相邻吸附氧直接偶联的贡献增加;ATR-SEIRAS 中 *OOH 峰逐步减弱而 *O-*O 峰增强,进一步支持 AEM 向 OPM 的切换。排除性验证:TMA+ 探针实验显示没有典型 LOM 中过氧样物种的响应,说明本文所称 OPM 不是晶格氧机制,而是发生在表面相邻 Fe 位点之间的 *O-*O 偶联路径。这个区分对于解释稳定性非常关键。性能闭环:HDFe-N-C 在三电极体系中 500 mA cm-2 仅需 288 mV 过电位,远低于商业 RuO2;在恒电流 OER 中可运行 2000 h;在 AEMWE 中 5 A cm-2 仅需 2.15 V 并稳定 500 h,还能经受 35000 次 0 到 5 A cm-2 启停循环。研究背景:文章面向阴离子交换膜水电解 AEMWE 的阳极析氧反应。AEMWE 兼具碱性体系低成本和膜电解高电流密度潜力,但阳极 OER 催化剂在高电流密度下同时需要高活性、低成本和长期稳定。存在的挑战/问题:当前先进催化剂在工业级动态工况下稳定性不足,并且对 OER 路径的精确控制有限。对 Fe-N-C 单原子体系而言,低金属负载和孤立位点通常导致 AEM 路径受线性标度关系约束。研究方案:作者通过理论计算和实验合成相结合,逐步提高 Fe-N-C 中 Fe 位点密度,并考察 Fe 负载对 *OH 覆盖、中间体谱学信号、同位素氧释放和 OER/AEMWE 性能的影响。核心亮点与发现:第一,高密度 Fe 位点可连续调控关键中间体吸附,尤其增强 *OH 覆盖;第二,Fe 负载升高使 OER 由 AEM 向 OPM 转换;第三,HDFe-N-C 在 14.7 wt.% Fe 负载下仍保持 Fe-N4 单原子分散;第四,该机制转换带来高电流 OER 和 AEMWE 的长周期稳定。研究意义与展望:文章提出“通过位点密度工程调控反应路径”的单原子催化剂设计策略,为非贵金属 OER 催化剂在工业水电解中的应用提供了可复制思路。亮点一:把 Fe 位点密度作为反应机制开关。论文并未停留在增加活性位数量,而是证明位点密度增加会改变 OER 的中间体覆盖与 O-O 键形成方式。亮点二:实现 14.7 wt.% 的高 Fe 单原子负载。高负载通常容易聚集成颗粒,本文通过高氮掺杂辅助策略和多尺度表征证明 Fe 仍主要以 Fe-N4 单原子形式存在。亮点三:用多种原位/操作态证据建立机制闭环。MOR、EIS、DEMS、ATR-SEIRAS 和 XAS 分别从吸附、界面动力学、同位素产物、振动峰和配位结构角度支持 OPM。亮点四:把材料性能推进到器件级。AEMWE 在 5 A cm-2 下 2.15 V、500 h 稳定运行,比单纯三电极数据更接近工业评价。亮点五:稳定性解释具有机制基础。OPM 不依赖晶格氧参与,理论上比 LOM 更不易引发结构流失;长期电解和启停循环数据与这一判断一致。前言首先确立应用场景:AEMWE 是连接低成本碱性电解和高电流密度膜电解的重要技术,但其工业化受限于阳极 OER 的慢动力学和高电流条件下的稳定性。随后作者把问题收窄到单原子催化剂:M-N-C SACs 具有位点清晰和原子利用率高的优势,却因为金属负载低,在实际电解槽中常需要较厚催化层,进而增加传质阻力和过电位。第二层逻辑是机制限制。低密度单原子位点彼此孤立,OER 通常沿 AEM 进行,即在同一位点上经历 *OH、*O、*OOH 等中间体逐步转化。这一路径受线性标度关系约束,难以独立优化各中间体吸附能。作者因此引入 OPM:在足够近的相邻金属位点上,两个 *O 物种可以直接偶联形成 O-O 键,从而绕开传统 AEM 的部分限制。第三层逻辑是材料可行性。二维氮掺杂碳支撑的单原子体系比刚性三维氧化物更容易调控局部结构和位点间距,因此高密度 Fe-N-C 可能既保持原子级分散,又提供相邻活性位协同。前言由此自然导向本文的核心命题:通过高密度 Fe 单原子位点,把 OER 路径从 AEM 定向切换到 OPM,并将这种机制优势转化为工业级 AEMWE 稳定性。Figure 1:Fe 位点密度驱动 OER 从 AEM 向 OPM 转换的理论框架。Figure 1 是全文的概念起点。低密度 Fe-N-C 中,活性位点彼此分离,OER 主要走 AEM,表面 *OH 覆盖度低,O-O 键形成依赖 *OOH 中间体;高密度 HDFe-N-C 中,相邻 Fe 位点增多,表面 *OH 富集并进一步形成相邻 *O,促发 *O-*O 偶联。图中的能量对比显示,HDFe-N-C 上 OPM 的速控步自由能低于低密度体系的 AEM 路径,说明位点密度改变的不只是活性位数量,也改变了反应路径与能垒。Figure 2:HDFe-N-C 的合成策略、原子分散与 Fe-N4 配位结构表征。Figure 2 解决“高密度是否还能保持单原子”这一关键前提。合成示意图强调高氮掺杂辅助策略可抑制 Fe 迁移和烧结;HAADF-STEM 中大量亮点对应原子级 Fe 物种,EDS 显示 Fe、N、C 分布均匀;ICP-OES 给出 14.7 wt.% Fe 负载。XANES/EXAFS 显示 Fe 处于正价态并以 Fe-N 为主配位,缺少 Fe-Fe 或 Fe-O-Fe 特征峰,WT-EXAFS 也支持 Fe-N 局域结构。这些数据共同证明材料不是 Fe 纳米颗粒或氧化铁,而是高密度 Fe-N4 单原子位点。Figure 3:不同 Fe 负载下 OER 机制从 AEM 到 OPM 的实验验证。Figure 3 是机制证据的核心。MOR 测试显示 Fe 负载越高,*OH 覆盖越强;operando EIS 显示与氧中间体吸附相关的低频响应增强;18O 标记 DEMS 中 36O2 信号随 Fe 含量升高而增加,说明相邻吸附氧直接偶联生成氧气的比例提升。ATR-SEIRAS 进一步显示,低 Fe 负载样品主要出现 *OOH 信号,高 Fe 负载样品则以 *O-*O 桥联中间体信号为主。最后的三维关联图把 Fe 含量、*OH 覆盖度和 OPM 比例连成线性关系,是全文最直接的“密度-覆盖-路径”证据。Figure 4:HDFe-N-C 的 OER 活性、长期稳定性和 AEMWE 器件性能。Figure 4 将机制优势转化为性能结果。HDFe-N-C 的 OER 极化曲线明显优于商业 RuO2 和 N-C,500 mA cm-2 时过电位仅 288 mV;质量活性、ECSA 归一化活性和 TOF 均显示单位位点活性更高。稳定性方面,HDFe-N-C 在 500 mA cm-2 下运行 2000 h,而商业 RuO2 快速衰减。器件层面,HDFe-N-C 作为 AEMWE 阳极可在 60 摄氏度下以 2.15 V 达到 5 A cm-2,并在 30 摄氏度、5 A cm-2 条件下稳定 500 h,说明该催化剂不是只在半电池中表现优异,而具备向工业电解槽迁移的潜力。本文建立了一个清晰的设计原则:当单原子位点从“稀疏、孤立”走向“高密度、相邻协同”时,催化剂可能从传统 AEM 反应范式中跳出,转向更有利的 *O-*O 偶联路径。对 OER 而言,这意味着单原子催化剂的优化不应只关注单个金属中心的配位环境,也应系统考虑位点密度、位点间距和表面覆盖度。未来值得进一步追问的问题包括:高密度 Fe-N4 位点的真实空间分布是否存在最优距离窗口;不同过渡金属或双金属 M-N-C 是否也能通过类似策略触发 OPM;在真实工业电解槽中,膜、离聚物、电极孔结构和气泡管理会如何影响 OPM 优势的发挥。若这些问题被继续推进,本文的机制框架可能扩展为一类面向工业电解的位点密度工程方法。本文解析基于用户提供的 PDF:Customized O-O Radical Coupling Route on High-Density Fe-N-C Catalysts for Stable Industrial-Scale Water Oxidation, Angewandte Chemie International Edition, 2026, DOI: 10.1002/anie.9504269。

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
