尽管二氧化钌(RuO2)在理论上具有比二氧化铱(IrO2)更优异的析氧反应(OER)活性和相对更低的价格,但平衡其活性与稳定性仍是一个重大挑战。Ru中心的电子结构在平衡RuO2基电催化剂的OER活性与耐久性方面起着关键作用。2026年6月8日,郑州大学王浩、卢思宇在Angewandte Chemie International Edition发表了题为"Fluorine-Doped RuO2 Anchored on TiO2 via Proton-Assisted Adsorption Evolution for Efficient and Stable Oxygen Evolution Reaction in Acid"的研究论文,Yaojia Cheng、Jingkun Yu为论文第一作者,王浩、卢思宇为论文通讯作者。
在本文中,作者开发了一种负载在TiO2上的F掺杂RuO2(F-RuO2@TiO2),构建了一个Ru-O-Ti界面平台,用于实现电负性介导的电荷重新分布。F-RuO2@TiO2表现出优异的OER活性和卓越的耐久性,在100和200 mA cm−2的电流密度下分别稳定运行了660小时和253小时。使用F-RuO2@TiO2组装的质子交换膜水电解器在0.5和1 A cm−2的电流密度下分别仅需1.57 V和1.68 V的电压,并分别稳定运行了300小时和100小时。
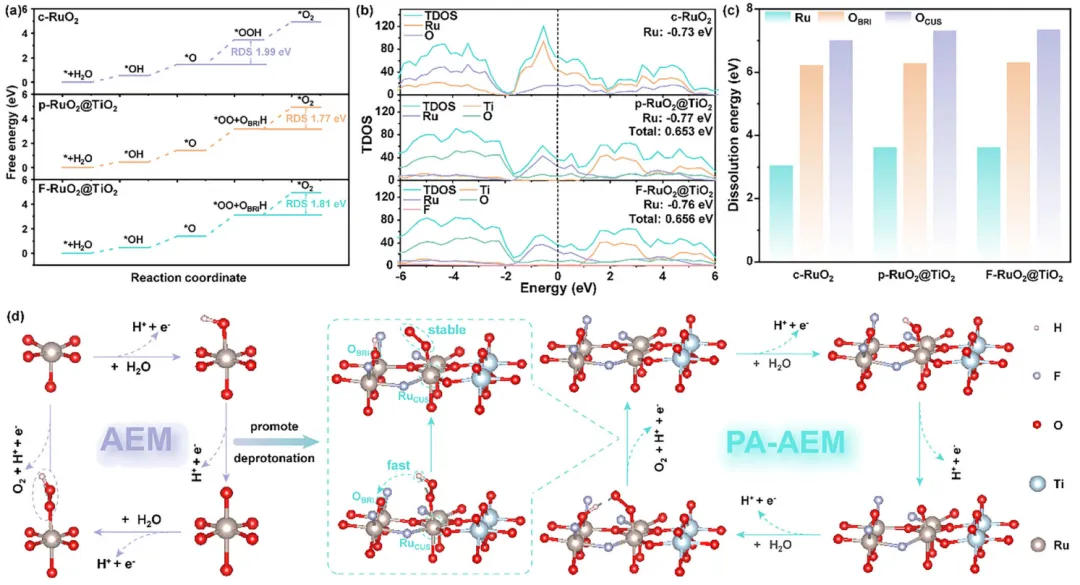
实验和密度泛函理论(DFT)计算均表明,F的高电负性增强了Ru-O共价性,从而通过质子辅助吸附演化机制(PA-AEM)加速了*OOH的去质子化过程。同时,Ru-O-Ti之间建立的动态电荷重新分布使得TiO2能够缓冲Ru位点处的电荷波动,从而进一步有效缓解过氧化。这些发现强调了电负性调控的质子转移动力学对于稳定RuO2的重要性。
酸性条件下的电催化析氧反应(OER)作为质子交换膜水电解器中的阳极反应,由于其在本质上缓慢的四电子转移和 O=O 键形成动力学,也是限制电解器制氢效率的关键反应。尽管贵金属 IrO2 基材料仍然是 PEMWEs 优异的商业化阳极催化剂,但其极低的丰度和较低的成本效益(约 140 美元/克)严重阻碍了 PEMWEs 的大规模开发和商业化。
研究人员发现,根据理论预测,RuO2和 Co3O4相比 IrO2表现出更优异的本征 OER 活性。然而,Co3O4在酸性条件下会快速溶解,这使得 RuO2基材料成为替代 IrO2作为 PEMWEs 阳极电催化剂的首选,从而引起了研究人员的广泛关注。尽管如此,RuO2基材料在高电压(> 1.4 V vs. RHE)的酸性环境中容易发生结构塌陷和溶解,因此无法长期用于 PEMWEs。
RuO2基电催化剂的基本局限性源于酸性 OER 过程中催化活性与结构耐久性之间固有的权衡关系,这构成了一个内在的热力学障碍。具体而言,遵循吸附演化机制的电催化剂由于 *OH 和 *OOH 中间体吸附能之间的比例关系而表现出较差的活性。在酸性环境中,高质子浓度会进一步加剧这一限制,因为它从热力学和动力学上抑制了 OOH 中间体中 O–H 键的断裂。
因此,OOH 的去质子化变得不利,阻碍了其向 *OO 物种的转化以及随后的 O2 释放,从而显著减慢了整体 OER 动力学。为了克服 AEM 途径的热力学限制,采用晶格氧介导途径的电催化剂可以通过引入晶格氧 OL来避免 *OH 和 *OOH 中间体的共存,打破它们之间的比例关系,从而提高电催化活性。此外,虽然 LOM 途径降低了对 *OOH 稳定化的依赖,但它并没有从根本上解决酸性条件下的质子转移限制。
相反,该反应涉及 OL的逃逸和再生,并且由于逃逸率远高于再生率,电催化剂结构容易发生塌陷,导致稳定性降低。因此,无论是 AEM 主导还是 LOM 主导的途径,在酸性 OER 中都存在固有的缺点,要么是由于与 *OOH 相关的质子转移效率低下,要么是由于过度的 OL参与,这凸显了开发能够同时促进质子转移同时保持结构完整性的替代策略的必要性。
除了对吸附能和 OL参与的传统考虑之外,最近的研究表明,促进 *OOH 中间体中质子的去除可以显著改善 OER 动力学。例如,阳离子的引入已被用于构建 Ru–O–M 界面单元,其中桥接氧 OBRI 原子可以促进 *OOH 物种中质子的去除。虽然这种策略可以有效促进 O–H 键的断裂并加速 OER 动力学,但对界面质子管理的进一步优化仍然至关重要。
然而,阳离子取代通常会导致可接触的 Ru 活性位点密度降低,并且经常伴随着引入的金属阳离子的溶解,从而部分牺牲了阳离子取代的 RuO2基电催化剂的活性和耐久性。最近的研究表明,用更具电负性的元素进行取代可以激活表面氧原子作为质子受体,从而稳定 OO 和 *H 中间体,并显著增强催化活性。值得注意的是,与阳离子取代不同,对 RuO2中氧原子进行阴离子取代不会降低可接触的 Ru 活性位点密度,这为提高催化性能而不牺牲活性位点密度提供了一种有效的策略。
基于上述考虑,作者成功合成了负载在 TiO2上的 F 掺杂 RuO2(F-RuO2@TiO2)电催化剂,该催化剂遵循质子辅助吸附演化机制途径。Ru–O–Ti 微框架可以激活邻近的 OBRI 原子作为质子受体,有效降低速率决定步骤的动力学能垒,并提高酸性 OER 活性。这种 RDS 动力学能垒的降低最终提高了酸性环境中 OER 的活性。
其中,TiO2的引入增强了 Ru 和 Ti 之间的电子相互作用,从而促进了高效的电荷转移并提高了电催化活性。高电负性 F 的引入增强了 Ru–O 共价键的稳定性,抑制了 Ru 和 OL的溶解,从而提高了电催化剂的稳定性。因此,电催化剂在酸性条件下的性能得到了显著提升。结果表明,F-RuO2@TiO2电催化剂在 10、50、100 和 200 mA cm-2的电流密度下分别表现出 170、207、224 和 236 mV 的低过电位。
这使得该电催化剂具有优异的长期稳定性(在 100 mA cm-2下稳定运行 660 小时,在 200 mA cm-2下稳定运行 253 小时)。以 F-RuO2@TiO2作为阳极电催化剂组装的 PEMWE,达到 0.5 和 1 A cm-2的电流密度分别仅需 1.57 V 和 1.68 V,并且可以分别稳定运行 300 小时和 100 小时。该工作通过促进界面质子转移来规避酸性条件下 OER 中 AEM 和 LOM 的固有限制,为合理设计 RuO2基电催化剂建立了一种实用的策略。
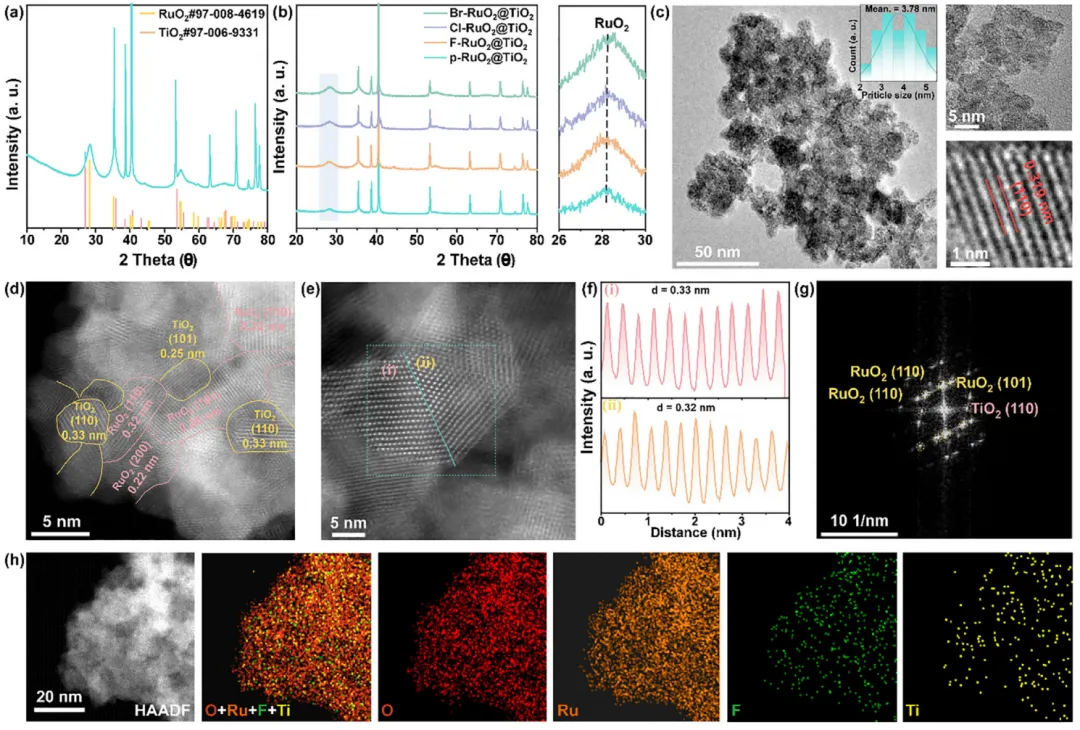
图1:X-RuO2@TiO2(X = F、Cl、Br)和 p-RuO2@TiO2的晶体结构与微观形貌。 (a) 以慢扫描速率(0.2 ° min-1)采集的 F-RuO2@TiO2的XRD图谱。(b) X-RuO2@TiO2和 p-RuO2@TiO2的 XRD 图谱。(c) F-RuO2@TiO2的TEM图像及相应的粒径分布图。(d, e) F-RuO2@TiO2的像差校正高角环形暗场扫描透射电子显微镜图像,显示 RuO2/TiO2异质界面。(f) 从 (e) 中标记区域提取的晶格条纹,分别对应于间距 0.33 nm 的 TiO2(110)晶面和间距 0.32 nm 的 RuO2(110)晶面。(g) (e) 所示区域的快速傅里叶变换(FFT)花样。(h) F-RuO2@TiO2的HAADF-STEM 图像及相应的 O、Ru、F 和 Ti 的 EDX 元素面分布图。
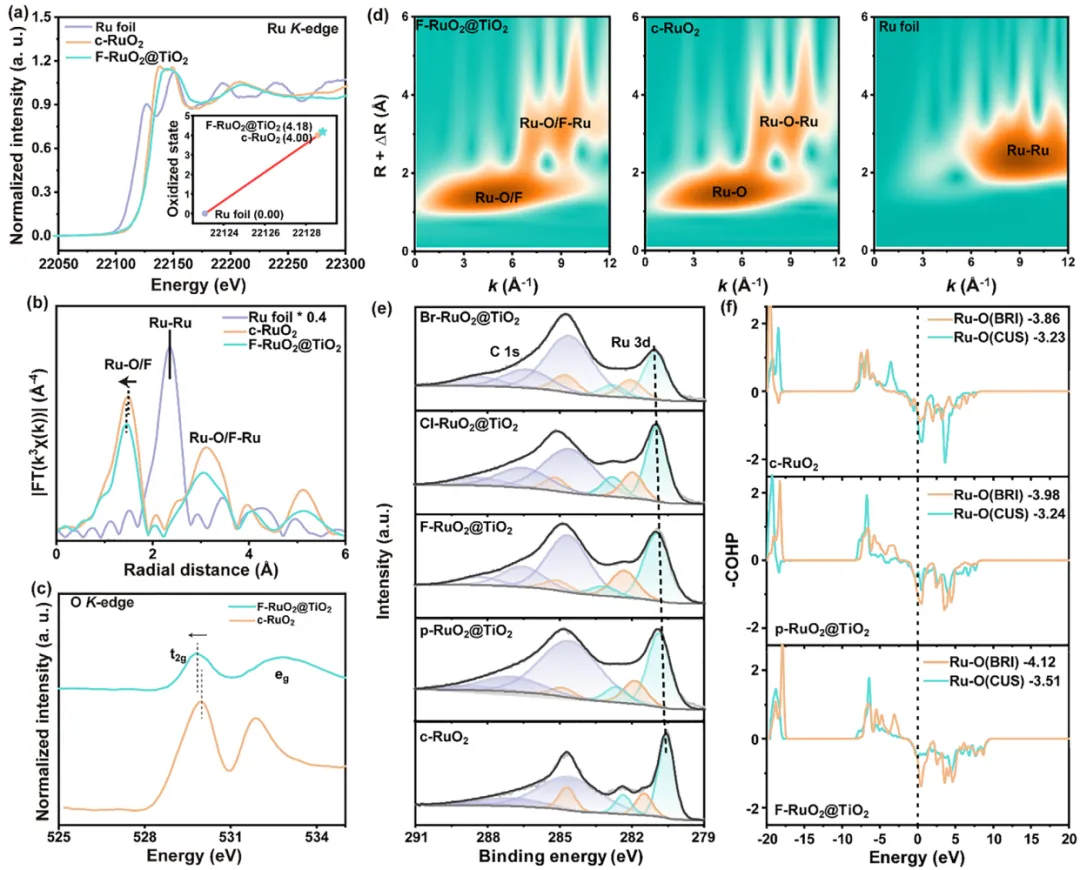
图2:所开发的RuO2基氧化物的电子结构与价态。 (a) Ru箔、F-RuO2@TiO2和c-RuO2的Ru K-edge光谱(插图为Ru原子的价态,以Ru箔和c-RuO2为标准参考)。(b) Ru箔、F-RuO2@TiO2和c-RuO2的Ru边FT-EXAFS光谱。(c) F-RuO2@TiO2和c-RuO2的O L-edge光谱。(d) Ru箔、F-RuO2@TiO2和c-RuO2在Ru K-edge的WT-EXAFS谱图。(e) F-RuO2@TiO2、Cl-RuO2@TiO2、Br-RuO2@TiO2、p-RuO2@TiO2和c-RuO2的C 1s + Ru 3d XPS光谱。(f) 通过DFT计算得到的c-RuO2、p-RuO2@TiO2和F-RuO2@TiO2中Ru–O键的COHP图。
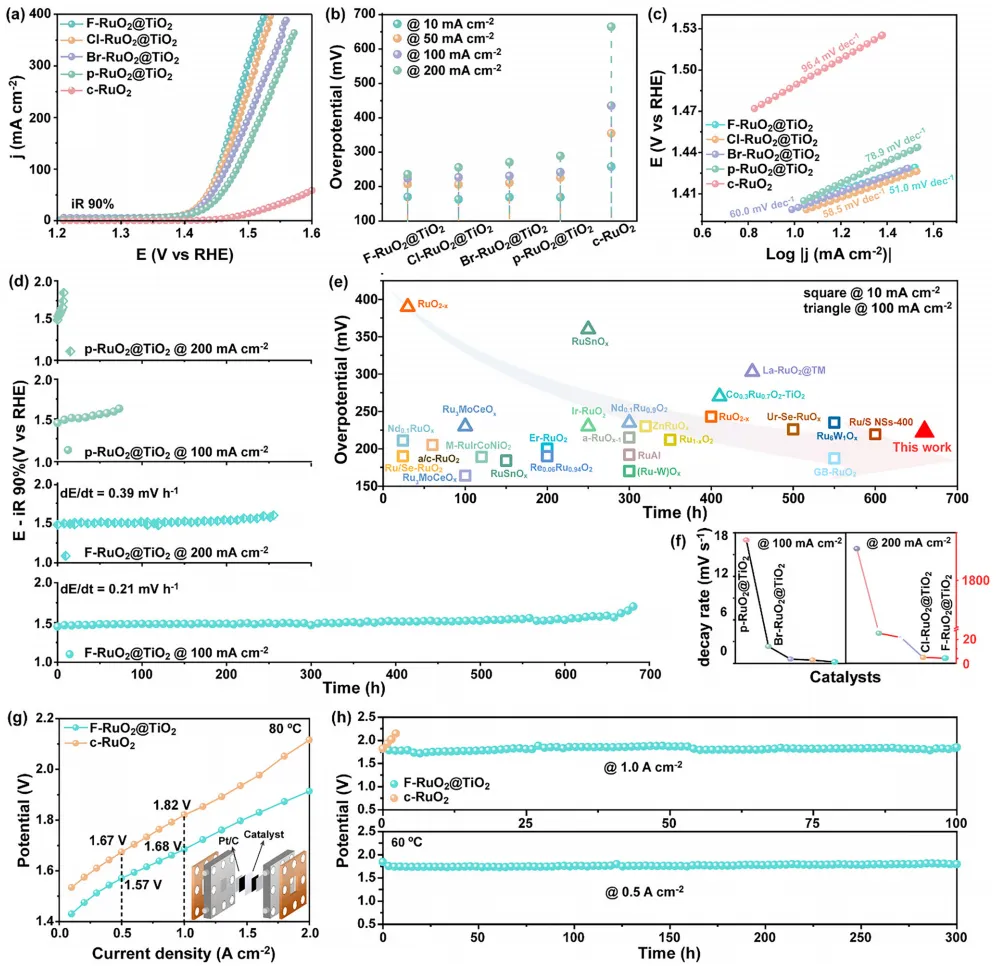
图3:X-RuO2@TiO2、p-RuO2@TiO2和c-RuO2在0.5 M H2SO4中的OER性能以及F-RuO2@TiO2在纯水中的PEMWE性能。 (a) 扫速为5 mV s-1、90% iR补偿下的LSV曲线。(b) 达到10、50、100和200 mA cm-2电流密度所需过电位的比较。(c) 由LSV曲线推导出的相应Tafel图。(d) F-RuO2@TiO2和p-RuO2@TiO2在100和200 mA cm-2恒电流密度下记录的CP曲线。(e) 活性-稳定性基准图,比较了F-RuO2@TiO2与文献中报道的代表性RuO2基酸性OER电催化剂在10和100 mA cm-2下的过电位及相应的稳定运行时间。(f) 从长期稳定性测试中提取的电压衰减率比较(左:100 mA cm-2,右:200 mA cm-2)。(g) 以F-RuO2@TiO2和c-RuO2为阳极组装的PEMWE的电流-电压极化曲线(插图为PEMWE示意图)。(h) 以F-RuO2@TiO2为阳极的PEMWE在60°C、0.5 A cm-2和1.0 A cm-2下的CP曲线。
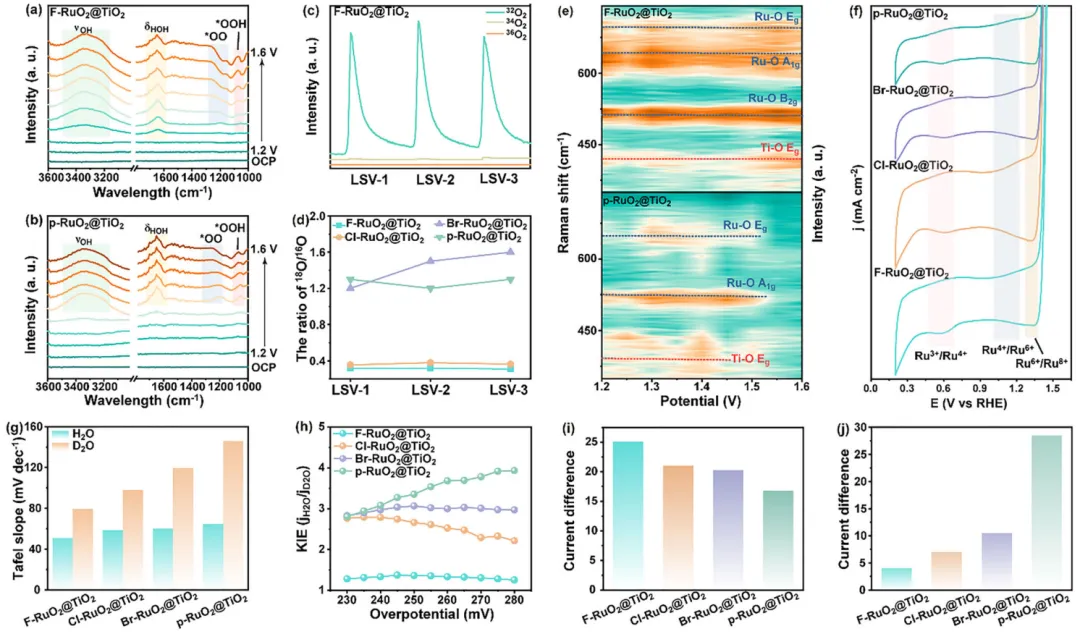
图4:通过原位表征和三电极体系分析OER机理。 (a) F-RuO2@TiO2和(b) p-RuO2@TiO2的原位SE-IR光谱。(c) F-RuO2@TiO2在0.5 M H2SO4中标记16O的DEMS数据。(d) 从DEMS数据得到的合成电催化剂的18O/16O比值。(e) F-RuO2@TiO2(上)和p-RuO2@TiO2(下)的原位Raman等高线数据图。(f) 合成电催化剂在0.5 M H2SO4中、0.2 V至1.4 V(vs. RHE)范围内的CV曲线。(g) 合成电催化剂在H2O或D2O的0.5 M H2SO4中的Tafel值。(h) 合成电催化剂在不同电位下获得的KIE(jH2O/jD2O)值。(i) 合成电催化剂在0.5 M H2SO4电解液中有无1.0 M CH3OH时LSV曲线之间的电流差。(j) 合成电催化剂在0.5 M H2SO4电解液中有无0.5 M TMA+时LSV曲线之间的电流差。
图5:DFT计算阐明了从传统AEM到PA-AEM途径的转变。 (a) c-RuO2、p-RuO2@TiO2和F-RuO2@TiO2在U = 0 V时的自由能图。(b) c-RuO2、p-RuO2@TiO2和F-RuO2@TiO2的TDOS结果。(c) Ru原子和O原子在CUS和BRI位点的溶解能。(d) 从传统AEM途径(c-RuO2)到PA-AEM途径(F-RuO2@TiO2)转变的示意图。
综上,该研究成功开发了一种氟掺杂二氧化钌负载二氧化钛(F-RuO2@TiO2)电催化剂,通过在Ru-O-Ti界面构建电负性介导的电荷重新分布机制,巧妙解决了RuO2基催化剂在酸性析氧反应中“活性高、稳定性差”的长期难题。实验表明,该催化剂在100 mA cm-2下可稳定运行660小时,在200 mA cm-2下可稳定运行253小时,远超目前多数RuO2基催化剂的耐久性水平。
研究团队提出了质子辅助吸附演化机制(PA-AEM)。氟的高电负性增强了Ru-O共价键的强度,并激活邻近的桥接氧原子作为质子受体,显著加速了*OOH中间体的去质子化过程,从而降低了反应速率决定步骤的能垒。同时,TiO2载体通过动态电荷重新分布缓冲了Ru位点的电荷波动,有效抑制了Ru的过氧化和溶解。这种“活性-稳定性协同优化”的策略,为突破传统AEM和LOM路径的固有限制提供了全新思路。
基于F-RuO2@TiO2组装的质子交换膜水电解器,在0.5 A cm-2和1.0 A cm-2下分别仅需1.57 V和1.68 V的电压,并可持续稳定运行300小时和100小时,展现出良好的实际应用潜力。未来,这一电负性调控质子转移的设计理念有望推广至其他酸性电催化体系,为低成本、高效率、长寿命的质子交换膜电解器阳极催化剂开发提供重要的理论指导和材料基础。
Fluorine-Doped RuO2 Anchored on TiO2 via Proton-Assisted Adsorption Evolution for Efficient and Stable Oxygen Evolution Reaction in Acid, Advanced Functional Materials, 2026, https://doi.org/10.1002/anie.9750153
#电催化#析氧反应#质子交换膜电解水#酸性OER#催化剂设计#氟掺杂#Angew#郑州大学#顶刊解读