「上海岱算科技有限公司」已向境内外230余家高等院校/科研院所提供了累计1400多项模拟计算服务,赋能科学研究提速增效!合作实验课题组在线发表学术论文期刊有ACS系列、AM系列、Angew、CEJ、EST、JACS、Matter、Nature子刊等,助力科研工作锦上添花!碱性析氧反应(OER)缓慢动力学,以及工业级安培级电流密度下的强极化条件所导致的催化剂不稳定性,促使研究人员探索新方法来打破具有挑战性的活性-稳定性权衡。
2026年06月12日,郑州大学卢思宇、常江伟团队在Angewandte Chemie International Edition期刊发表题为“Ultrastable Non-Noble-Metal Oxygen Evolution Electrocatalyst for Industrial-Level Water Electrolysis”的研究论文,团队成员胡志昂、于镜坤为论文共同第一作者,卢思宇、常江伟为论文共同通讯作者。
第一作者:胡志昂、于镜坤
通讯作者:卢思宇、常江伟
通讯单位:郑州大学
论文DOI:10.1002/anie.2311940
该研究证明,向Fe₂O₃超薄纳米片八面体空隙中进行原子级碳掺杂(C-Fe₂O₃UNSs),构建了Feₒₕᴵᴵᴵ–O–Feₒₕᴵᴵᴵ协同中心,可以调控*OH中间体的覆盖度,并允许直接*O–*O自由基耦合。这同时实现了结构与催化稳定性,原位光谱测量和密度泛函理论DFT计算证实了这一点,该催化剂达到500mA cm⁻²的过电位为227mV,并具有4500小时的耐久性。将C-Fe₂O₃UNSs应用于阴离子交换膜水电解槽中时,在1.5A cm⁻²电流密度下提供1.72V的电池电压,并在1~5A cm⁻²电流密度下运行2800小时,在3A cm⁻²电流密度下的衰减率为3.05mV kh⁻¹。
随着与化石燃料燃烧相关的环境问题日益严峻,开发更可持续的清洁能源技术的需求变得更加紧迫。长期以来,利用阴离子交换膜水电解槽生产绿氢一直被认为是制造绿氢燃料的一种途径。然而,AEMWE技术的实际应用目前受到阳极析氧反应(OER)动力学缓慢的阻碍。尽管某些基于RuO₂和IrO₂的贵金属催化剂已在AEMWE的安培级电流密度下表现出良好的活性,但要使AEMWE技术在成本和能效方面与质子交换膜水电解竞争,需要高效且耐用的非贵金属OER催化剂。
当前针对OER催化剂的研发工作主要集中在通过改变金属的配位环境来增强本征反应性,即提高转换频率(TOF),这依赖于两种通用策略:(i)基于吸附质演化机制(AEM),优化含氧中间体(如*OH、*O和*OOH)的吸附能。在这种情况下,鉴于*OH和*OOH中间体之间结合能的差异几乎恒定,可以看到一个线性标度关系,其OER理论过电位约为370mV;(ii)激活晶格氧(Olat)参与OER,即所谓的晶格氧介导机制(LOM)。基于LOM的OER过程不再仅由活性金属决定,而是通过阴离子氧化还原过程激活Olat并形成O⁽²⁻ᵟ⁾⁻配体(公众号:生化环材人)。因此,被激活并参与析氧的Olat会触发大量氧空位的形成,而体相氧的动态迁移以补充表面氧空位则会导致结构不稳定性。相应地,稳定OER电催化剂的主要策略是抑制LOM路径,尽管这是以牺牲活性为代价的。以牺牲活性换取稳定性是一种短期的权衡,并非开发卓越OER电催化剂的真正可行策略。
为了同时提高用于AEMWEs的OER催化剂的活性和耐久性,研究人员目前正在探索多种新方法。近期研究强调了调控电化学界面(如界面水结构和吸附氧物种)的重要性。原则上,电催化反应发生在电极与电解质之间的带电界面上,这个狭窄区域不仅影响电催化反应动力学,也为优化电催化性能提供了机会。通过控制OER活性位点的结构,可以在界面电解质微环境中优化吸附行为(例如,促进来自吸附水中的氧而非阳极氧化物晶格中氧的参与),从而抑制金属溶解并提高稳定性。更重要的是,近期研究提出了一种新策略,通过合理富集*OH覆盖度来促进*O─*O键的形成,从而激活氧化物路径机制(OPM)路径。理论上,OPM基电催化剂能够在没有Olat参与和*OOH生成的情况下实现*O自由基的直接耦合,从根本上打破了活性与稳定性之间的传统权衡关系。要触发OPM,需要对催化剂的几何结构和电子结构进行精确控制,以实现OER中间体的最佳吸附强度/几何构型,而这一点迄今为止在实验上仍具有挑战性。这促使研究人员共同解决催化剂结构和含氧物种吸附状态的问题,以便在无贵金属的AEMWE阳极上实现OER过程中的OPM。
在此,该研究证明,在Fe₂O₃超薄纳米片(C-Fe₂O₃ UNSs)中的FeO₆八面体周围进行碳掺杂可以优化Feₒₕᴵᴵᴵ活性中心,从而调控OER过程中含氧中间体的吸附能和吸附物种,进而实现高活性和高稳定性AEMWE性能。具体而言,所构建的Feₒₕᴵᴵᴵ–O–C配体创造了具有上移Fe 3d轨道和降低的Feₒₕᴵᴵᴵ–O共价性的Feₒₕᴵᴵᴵ–O–Feₒₕᴵᴵᴵ位点,这首先增强了*OH吸附和电荷转移。实验和理论证明,这降低了Feₒₕᴵᴵᴵ–O–Feₒₕᴵᴵᴵ位点上*O偶联所需的能垒,有利于通过OPM路径在界面微环境中形成桥连*O–*O中间体,而不是通过AEM路径使*OH与预吸附的*O反应生成*OOH中间体(公众号:生化环材人)。研究结果表明,C-Fe₂O₃ UNSs表现出高活性和卓越的稳定性,达到500mA cm⁻²电流密度所需的超低过电位约为227mV,并具有超过4500小时的优异催化稳定性。此外,在1.5A cm⁻²的电流密度下,基于C-Fe₂O₃ UNSs的AEMWE可以实现1.72V的电池电压。在从1~5A cm⁻²的阶跃加速老化实验中,该电解器在2800小时内稳定运行,电压衰减率仅为3.05mV kh⁻¹,这与美国能源部(DOE)设定的目标相当。更重要的是,该研究提出的碳掺杂方法可以应用于各种单金属氧化物以及高熵过渡金属氧化物纳米片。这种方法在设计高性能多功能OER电催化剂方面的普适性,有望拓展过渡金属氧化物基材料在能源存储/转换系统中的应用。
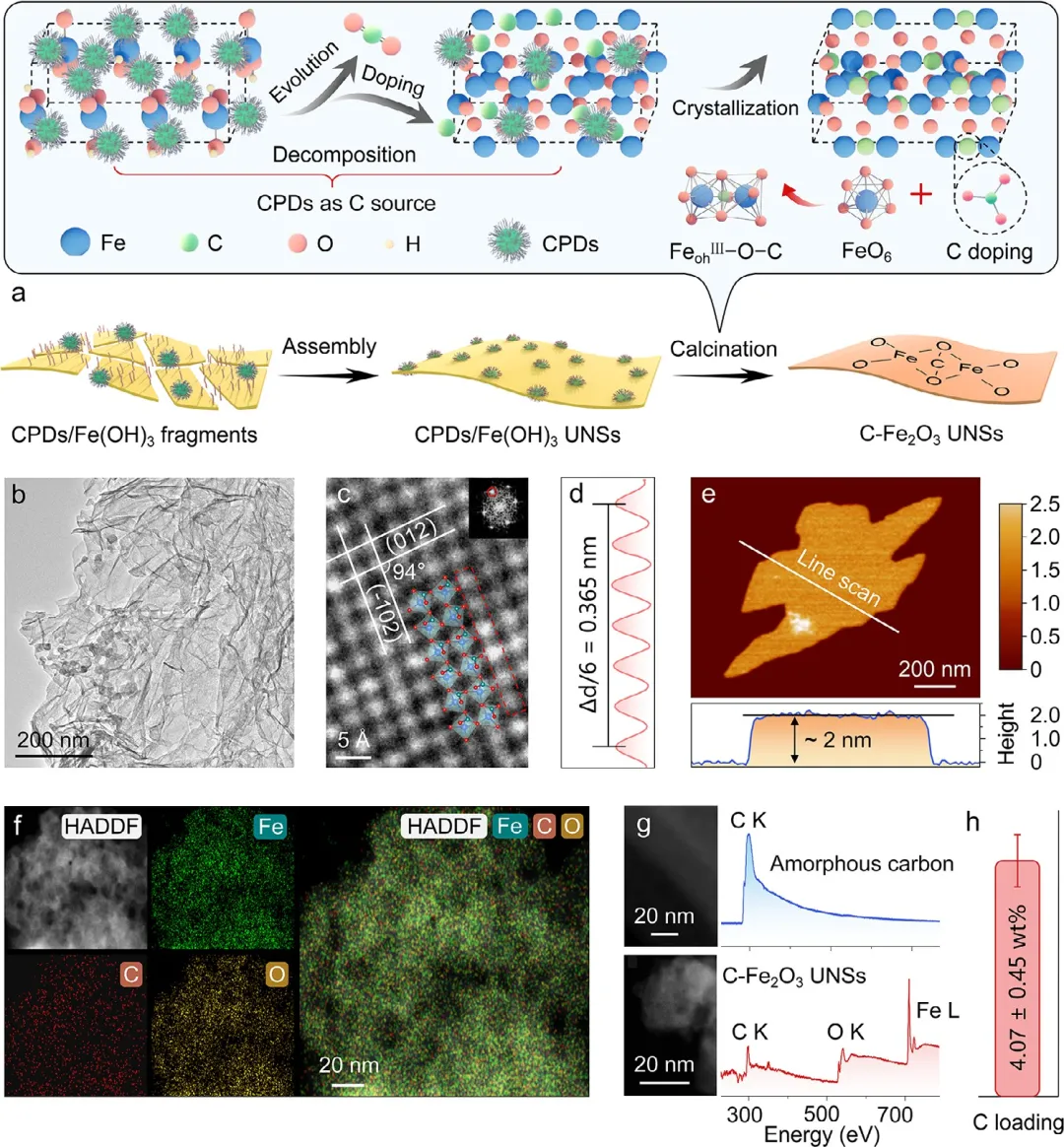
图1 | C-Fe₂O₃ UNSs的合成与结构表征。(a) 以CPDs作为碳源,在Fe₂O₃前驱体与CPDs同步热解过程中进行碳掺杂方法的示意图。(b) C-Fe₂O₃ UNSs的HAADF-STEM图像,显示出纳米片形貌。(c) C-Fe₂O₃ UNSs的高分辨率HAADF-STEM图像。插图为相应的FFT图。(d) (c)中红色虚线方框标记区域的线扫描强度分布图,显示出(012)晶面的晶面间距。(e) C-Fe₂O₃ UNSs的AFM图像(公众号:生化环材人)。(f) C-Fe₂O₃ UNSs的ADF图像以及Fe、O、C的局部能量色散X射线光谱元素分布图及其叠加图像,表明各元素均匀分散。(g) 使用高角环形暗场探测器采集的C-Fe₂O₃ UNSs的STEM图像,以及显示不同元素吸收边的相应电子能量损失谱:无定形碳(左上/右)和C-Fe₂O₃ UNSs(左下/右)。(h) C-Fe₂O₃ UNSs中的平均碳掺杂含量。
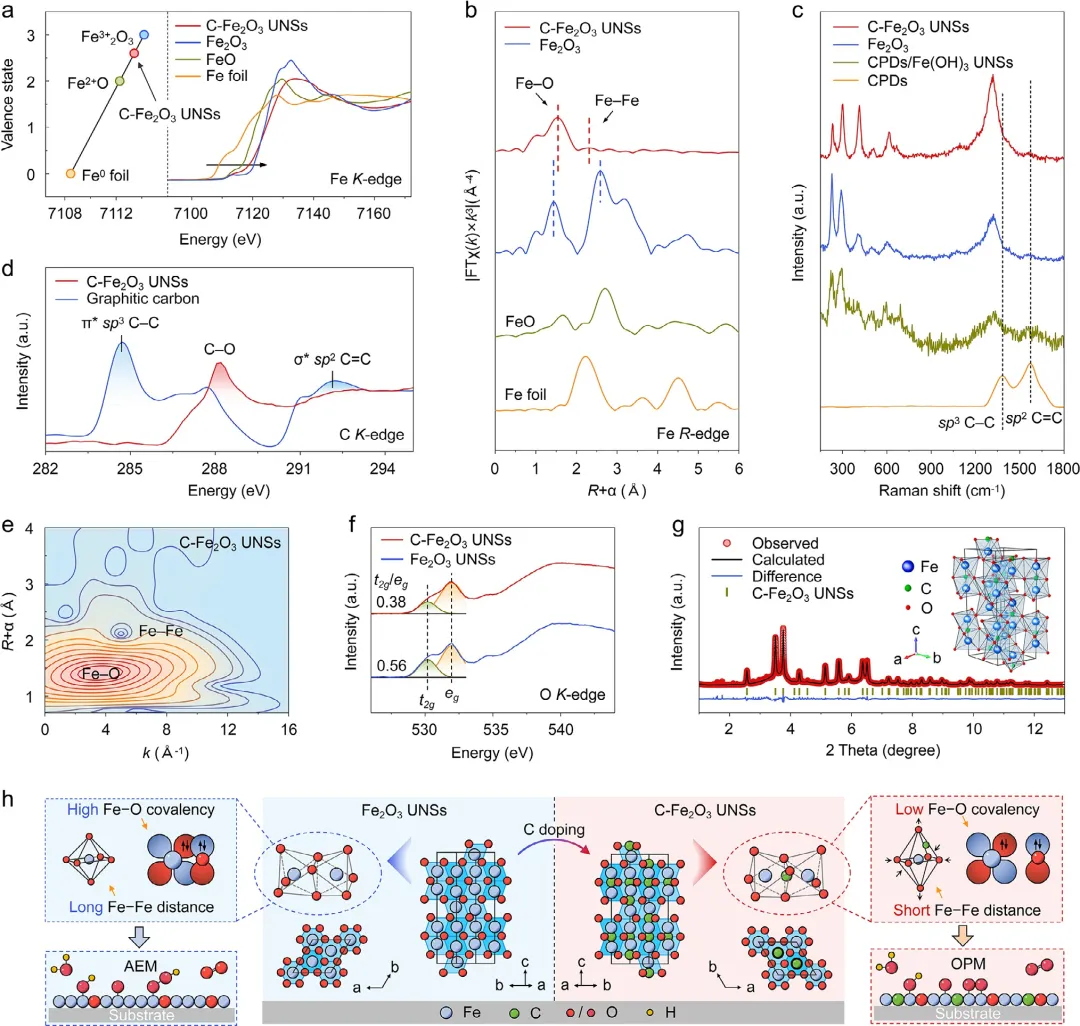
图2 | C-Fe₂O₃ UNSs的化学态与配位环境。(a) C-Fe₂O₃ UNSs及所选参比材料(包括Fe₂O₃、FeO和Fe箔)的归一化Fe K边XANES谱图。(b) C-Fe₂O₃ UNSs、商业Fe₂O₃、FeO和Fe箔的归一化傅里叶变换Fe K边EXAFS谱图。(c) 拉曼光谱。(d) C-Fe₂O₃ UNSs的C K边XANES谱,以石墨碳为参比。(e) C-Fe₂O₃ UNSs的k³加权WT-EXAFS等高线图。(f) C-Fe₂O₃ UNSs和Fe₂O₃ UNSs的O K边XANES谱(公众号:生化环材人)。拟合区域涉及O 1s电子向O 2p-Fe 3d杂化轨道的跃迁,其中绿色和黄色阴影区域分别对应于Fe 3d t2g和eg态。图中标出了两个样品的t2g/eg强度比。(g) C-Fe₂O₃ UNSs的SXRD图。(h) 通过碳掺杂在Feₒₕᴵᴵᴵ–O单元中实现电子和结构调控以允许OPM路径的示意图。
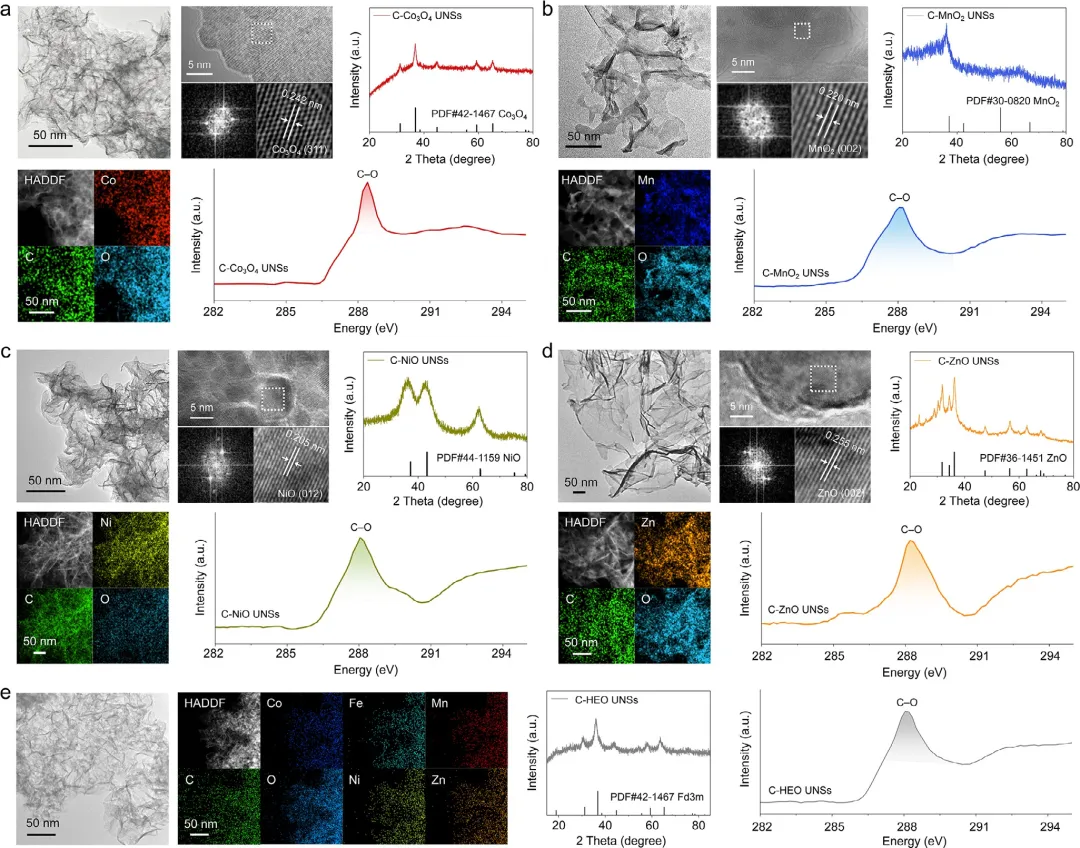
图3 | 原子级碳掺杂到其他金属氧化物纳米片的普适性。(a-d) (a) C-Co₃O₄ UNSs、(b) C-MnO₂ UNSs、(c) C-NiO UNSs和(d) C-ZnO UNSs的结构分析。左上:HAADF-STEM图像;左中:HRTEM图像、高亮区域的FFT图像及相应的反FFT图像;右上:XRD图谱;左下:EDS元素分布图;右下:C K边XANES谱。(e) 尖晶石型C-HEO UNSs的表征,其金属元素组成为Co、Mn、Fe、Ni和Zn。
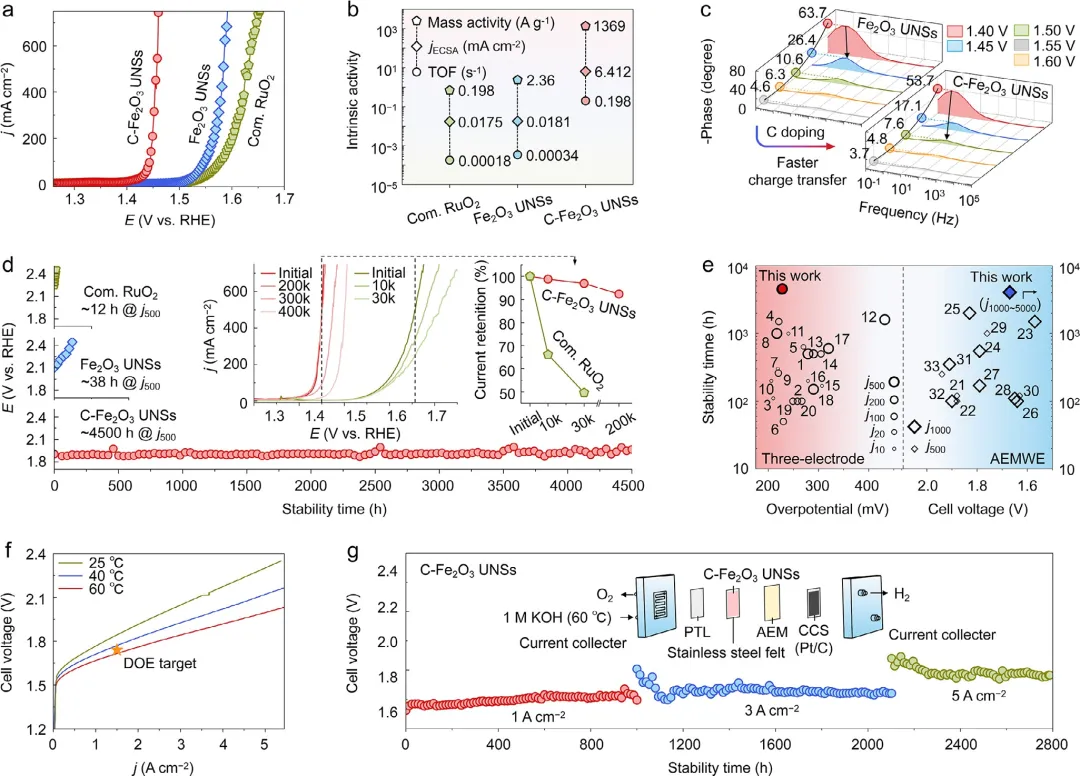
图4 | 电化学OER性能。(a) C-Fe₂O₃ UNSs、Fe₂O₃ UNSs和商业RuO₂在1.0M KOH中测得的LSV曲线。(b) 在1.457V电位下计算的本征活性比较。(c) C-Fe₂O₃ UNSs和Fe₂O₃ UNSs的原位电化学阻抗谱。(d) C-Fe₂O₃ UNSs、Fe₂O₃ UNSs和商业RuO₂在三电极系统中测得的计时电位曲线(公众号:生化环材人)。所有电压均未进行iR补偿。插图显示了加速伏安循环前后稳态析氧极化曲线。(e) C-Fe₂O₃ UNSs在AEMWE中的极化曲线。(f) 基于C-Fe₂O₃ UNSs阳极的AEMWE在1、3和5A cm⁻²顺序电流密度下的计时电位曲线。插图为AEMWE装置示意图。PTL:多孔传输层;AEM:阴离子交换膜;CCS:催化剂涂覆基底。(g) C-Fe₂O₃ UNSs的性能与其他近期报道的OER基准催化剂的比较。
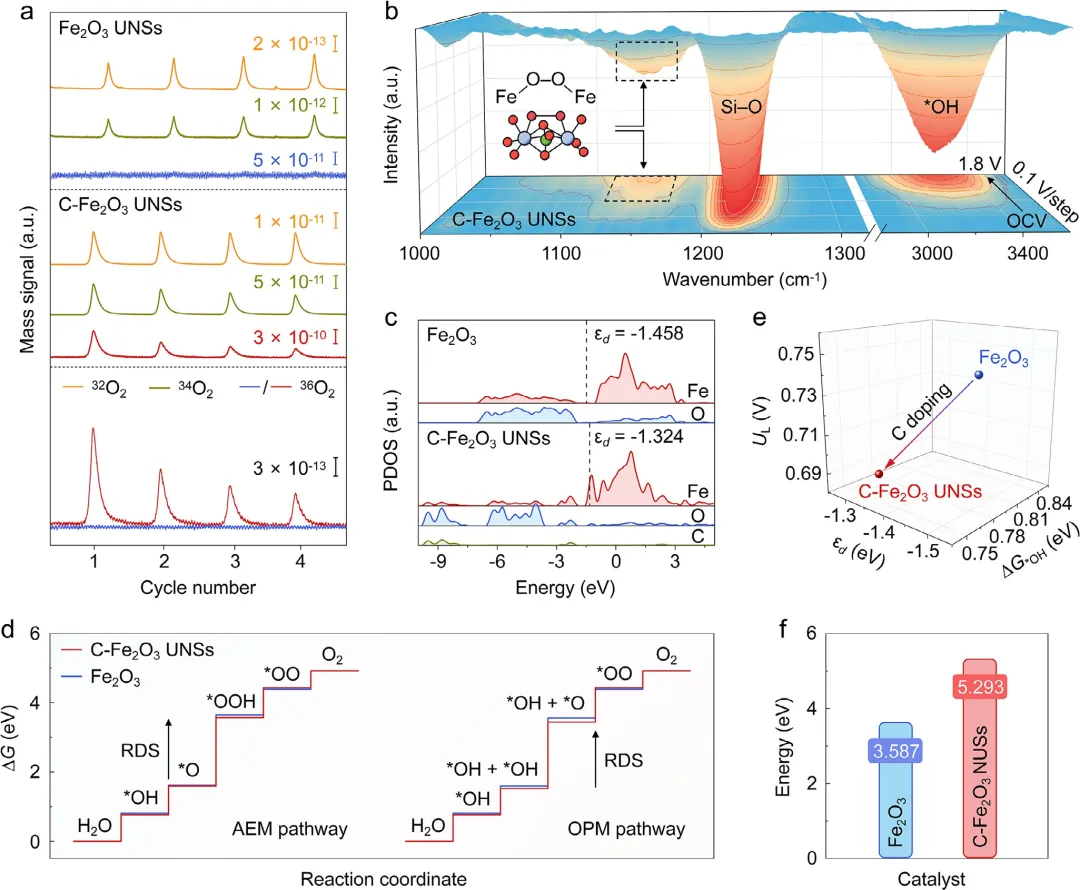
图5 | 原位机理研究与密度泛函理论DFT计算。(a) ¹⁸O标记的Fe₂O₃ UNSs(上)和¹⁸O标记的C-Fe₂O₃ UNSs(中)在使用H₂¹⁶O作为溶剂的1M KOH电解液中测试时析氧的原位微分电化学质谱信号,以及C-Fe₂O₃ UNSs和Fe₂O₃ UNSs之间³⁶O₂/³²O₂信号的详细比较(下)。(b) C-Fe₂O₃ UNSs和Fe₂O₃ UNSs的原位衰减全反射表面增强红外吸收光谱。(c) C-Fe₂O₃ UNSs和Fe₂O₃上Fe 3d轨道和O/C 2p轨道的分波态密度带中心(公众号:生化环材人)。(d) C-Fe₂O₃ UNSs和Fe₂O₃分别遵循AEM和OPM路径的ΔG图。(e) 基于OPM路径的C-Fe₂O₃ UNSs和Fe₂O₃上过电位作为ΔG*OH和εd函数的三维散点图。(f) C-Fe₂O₃ UNSs和Fe₂O₃的Fe脱金属化计算能。
总之,该研究报道了一种无贵金属C-Fe₂O₃ UNSs,其具有通过FeO₆八面体周围的碳掺杂调控的特定Feₒₕᴵᴵᴵ–O–Feₒₕᴵᴵᴵ构型,是一种超稳定的OER催化剂。广泛的原位电化学表征以及理论计算合理地解释了优化的Feₒₕᴵᴵᴵ中心能够(i)增强*O覆盖度,进而促进Feₒₕᴵᴵᴵ–O–Feₒₕᴵᴵᴵ单元*O–*O自由基耦合,从而触发低能垒OPM路径;(ii)由于Feₒₕᴵᴵᴵ–O共价性减弱而阻止了晶格氧的参与,从而同时实现了电催化和结构稳定性。值得注意的是,C-Fe₂O₃ UNSs在500mA cm⁻²电流密度下表现出227mV的低过电位,并可稳定电催化长达4500小时。此外,基于C-Fe₂O₃ UNSs阳极的AEMWE在1.5A cm⁻²电流密度下实现了1.72V的电压,并且在2800小时内从1到5A cm⁻²的顺序测试中显示出极低的衰减率(例如在3A cm⁻²下为3.05mV kh⁻¹),从而满足了AEMWE系统的严格DOE技术目标。该研究为用于水氧化的稳定非贵金属电催化剂开辟了一条有前景的途径,代表了碱性水电解槽规模化发展的重要进步。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
