视网膜色素变性(Retinitis Pigmentosa,RP)是导致遗传性失明的主要原因之一,目前尚缺乏有效治疗方法,因此亟需深入阐明其发病机制并寻找新的治疗靶点。本研究旨在通过整合多模态数据分析框架,揭示程序性细胞死亡(programmed cell death,PCD)相关基因在RP发病中的遗传学作用。本研究整合了全基因组关联研究(GWAS)、表达数量性状位点(eQTL)、蛋白质数量性状位点(pQTL)以及处理后的转录组数据。
研究流程包括:以双样本孟德尔随机化为主要因果推断方法;采用基于汇总数据的孟德尔随机化(summary-data-based MR,SMR)进行补充验证;利用贝叶斯共定位分析(Bayesian colocalization)、多变量孟德尔随机化(multivariable MR,MVMR)、全表型关联研究(phenome-wide association study,PheWAS)以及基于GTEx eQTL数据的组织特异性MR分析进行特异性验证;结合整体RNA测序(bulk RNA-seq)和单细胞RNA测序(single-cell RNA-seq)开展功能注释。本研究使用公开可获得的遗传学和转录组数据库。主要分析采用RP的GWAS汇总统计数据(ebi-a-GCST90018912、ebi-a-GCST90013904 和 ebi-a-GCST90013954)。
验证分析采用:bulk RNA-seq数据集:GSE62020;单细胞RNA-seq数据集:GSE183206。暴露因素为PCD相关基因的遗传工具变量化表达水平(cis-eQTL)或蛋白表达水平(cis-pQTL)。主要结局为视网膜色素变性(RP)。次要结局包括:基因表达异常;功能富集分析;免疫细胞浸润;细胞类型特异性表达。
结果
综合双样本MR和SMR分析,共鉴定出8个核心PCD相关基因,具有充分的遗传学证据支持其与RP发生相关。例如:AIM2 OR =2.90 95% CI:1.81–4.63上述基因在多个组织中均表现出一致的效应,并且在调整吸烟和体力活动等代理表型后的多变量MR(MVMR)分析中仍保持名义统计学显著性(nominal P < 0.05)。PheWAS分析未发现这些关联存在明显的多效性(所有P值均 > 5 × 10⁻⁸)。在视网膜转录组数据中,这些核心基因均存在异常表达,并显著富集于免疫相关通路。进一步利用CIBERSORT进行免疫浸润分析发现:多种免疫细胞浸润与这些基因密切相关;其中,H13与M1型巨噬细胞之间存在显著负相关:r = −0.625P < 0.05。单细胞RNA测序进一步显示:Müller胶质细胞是重编程程度最高的细胞群体(AUC评分 =0.07)
结论
本研究通过多模态整合分析,为一组核心程序性细胞死亡相关基因参与视网膜色素变性(RP)发病提供了遗传学证据。研究结果提示:这些基因的异常调控可能通过引发免疫稳态失衡以及Müller胶质细胞功能障碍,共同促进RP的发生和发展。上述发现揭示了RP潜在的发病机制,并为这一致盲性疾病提供了一组具有应用前景的治疗靶点网络。
关键词
视网膜色素变性;程序性细胞死亡相关基因;孟德尔随机化;表达数量性状位点;蛋白质数量性状位点图1:技术路线图
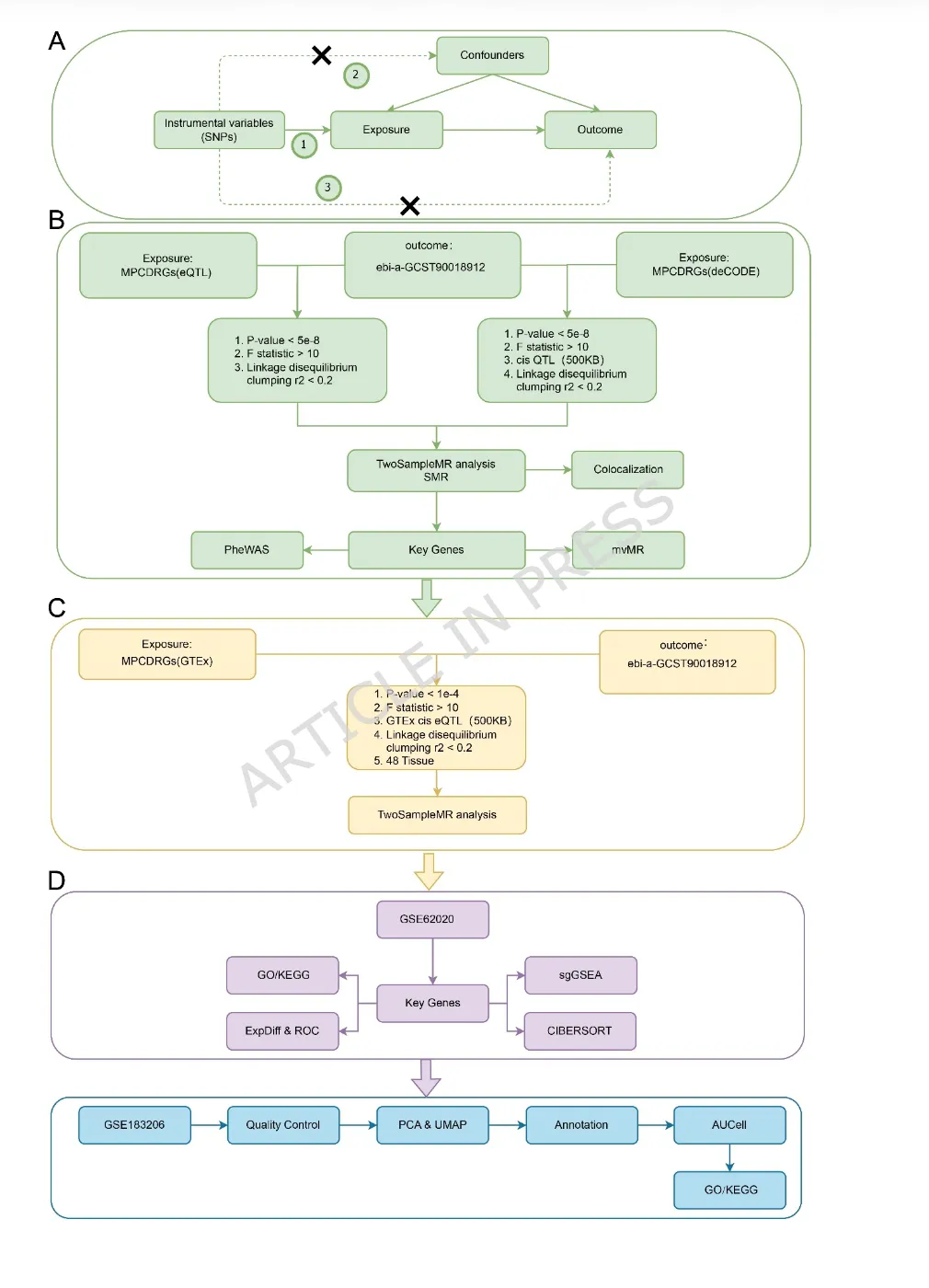
(A)孟德尔随机化推断成立的核心假设:1)相关性(Relevance):遗传工具变量与暴露因素具有稳健的关联;2)独立性(Independence):遗传工具变量与暴露–结局关系中的混杂因素无关联;3)排除限制(Exclusion restriction):遗传工具变量仅通过暴露因素影响结局,而不存在其他作用途径。(B)本研究开展孟德尔随机化分析的技术流程。(C)基于GTEx(Genotype-Tissue Expression)数据进行孟德尔随机化验证的技术流程。(D)基于GEO(Gene Expression Omnibus)公共数据库的批量转录组(bulk transcriptomic)和单细胞转录组(single-cell)数据进行基因表达验证的技术流程。
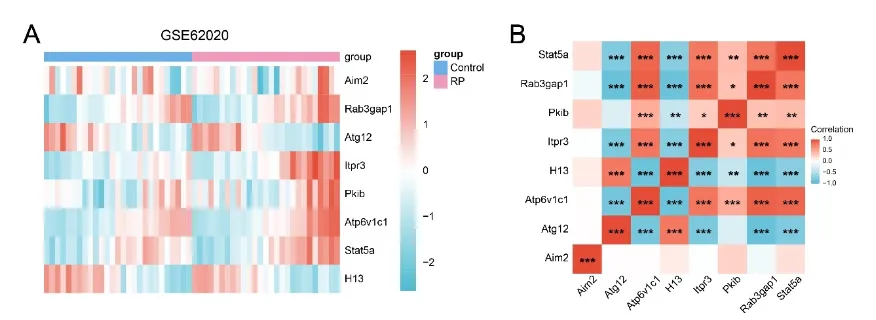
图2:视网膜色素变性关键基因的转录组表达特征及相关性分析
(A)显示八个关键基因表达谱的热图(蓝色:对照组;粉色:视网膜色素变性(Retinitis Pigmentosa,RP)组)。(B)显示八个关键基因表达水平之间相关性的热图。
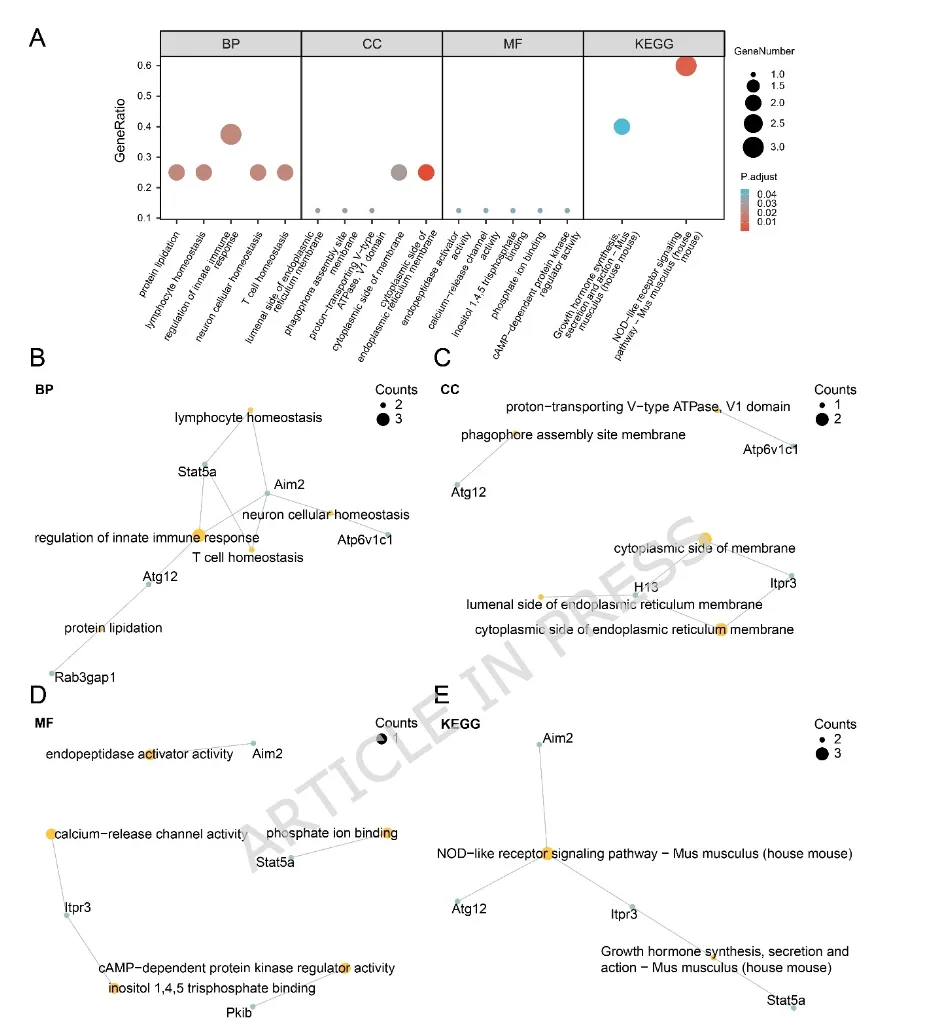
图3:关键基因的功能富集分析
(A)基因本体(Gene Ontology,GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析结果气泡图。富集条目分为生物过程(Biological Process,BP)、细胞组分(Cellular Component,CC)、分子功能(Molecular Function,MF)和KEGG通路。气泡大小表示富集基因数目,颜色表示校正后的P值。(B–E)关键基因与BP(B)、CC(C)、MF(D)及KEGG通路(E)富集条目之间关联关系的网络图。
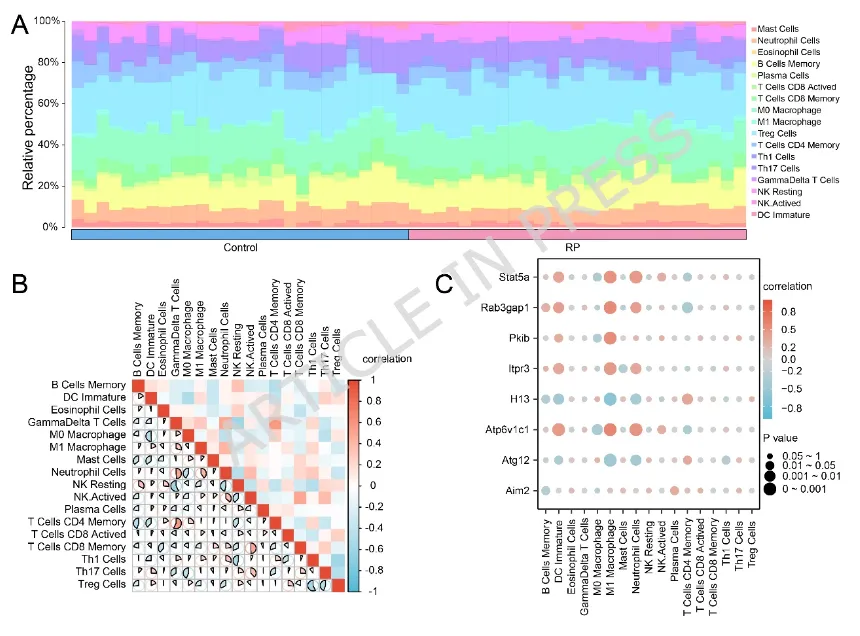
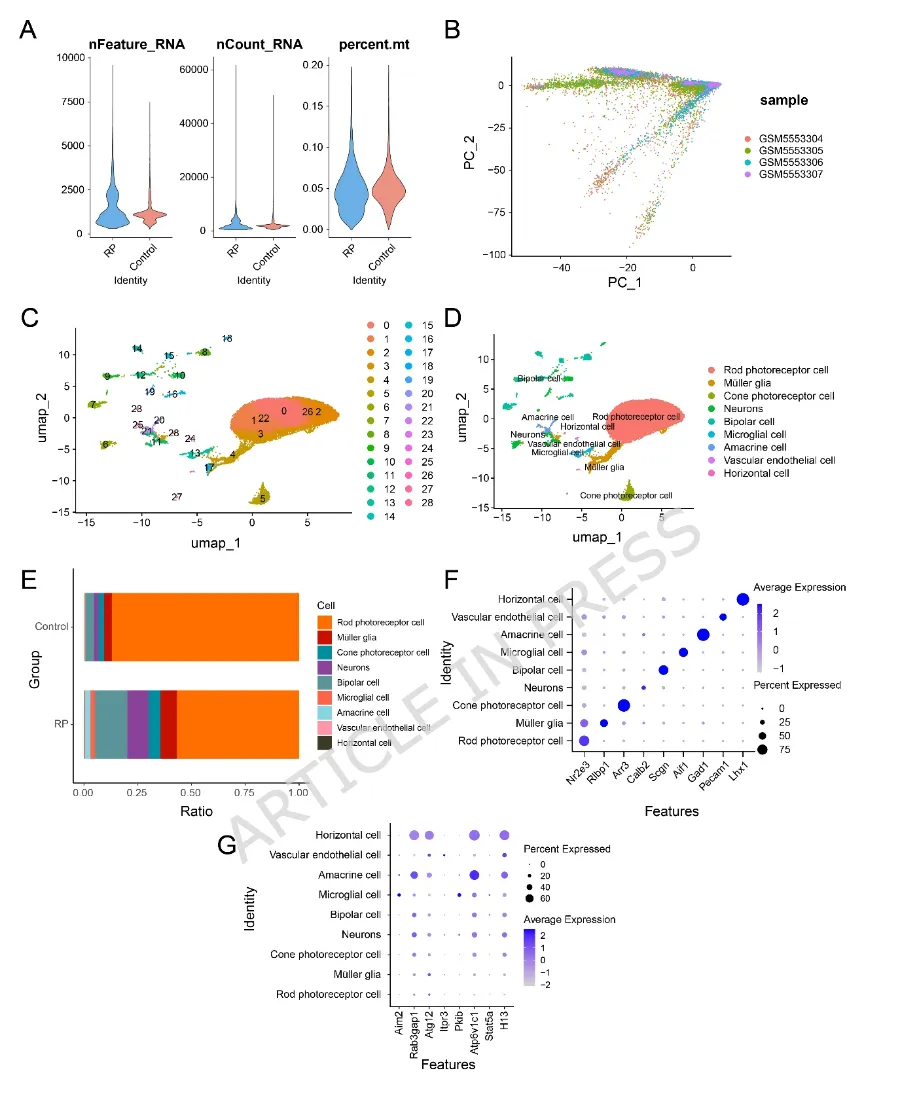
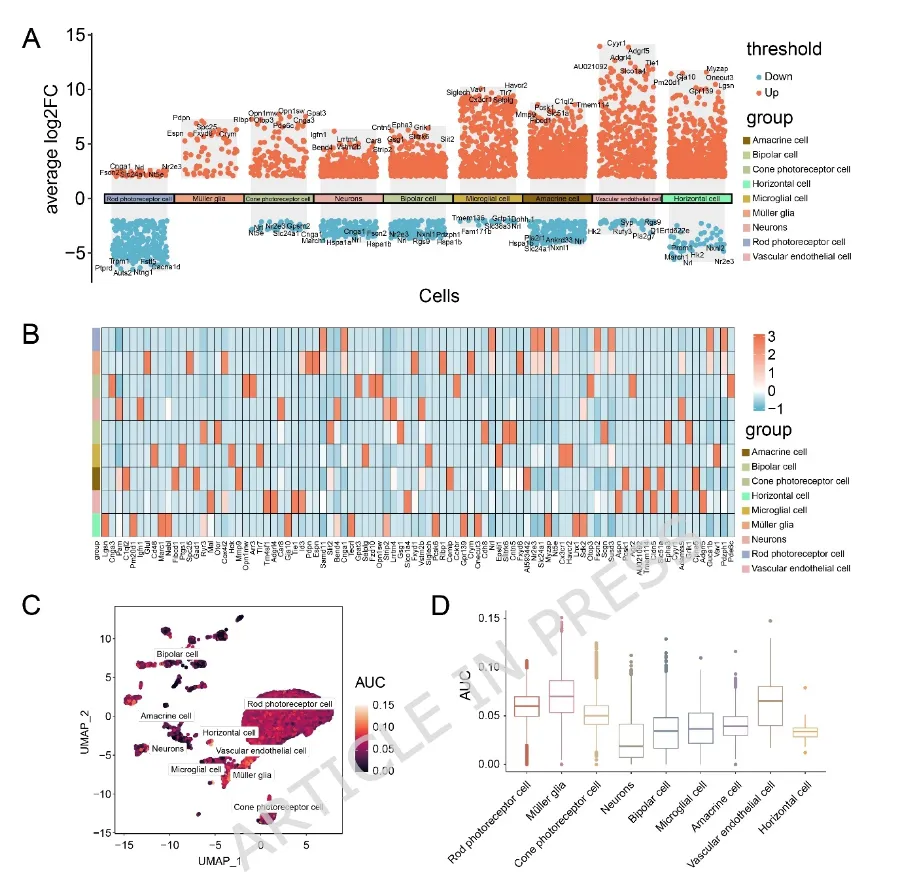
图4:(A)显示各样本中免疫细胞类型估计相对比例的柱状图(蓝色:对照组;粉色:视网膜色素变性(Retinitis Pigmentosa,RP)组)。(B)不同免疫细胞类型浸润水平两两相关性的热图。颜色条表示Pearson相关系数(r),取值范围为−1(蓝色,负相关)至1(红色,正相关)。(C)展示八个关键基因表达水平与免疫细胞浸润丰度相关性的气泡图。图5:(A)展示单细胞RNA测序数据三项关键质量控制指标的小提琴图。nFeature_RNA:反映转录复杂度(检测到的基因数目);nCount_RNA:反映测序深度(RNA总计数);percent.mt:反映细胞应激或损伤状态(线粒体基因所占百分比)。剔除nCount_RNA<500、nFeature_RNA<250或percent.mt>20%的低质量细胞后进行后续分析。(B)主成分分析(Principal Component Analysis,PCA)图,展示不同样本细胞的转录组特征。(C)统一流形近似与投影(Uniform Manifold Approximation and Projection,UMAP)图,展示通过无监督聚类识别得到的29个细胞簇。(D)UMAP图,展示细胞被注释为9种主要细胞类型。(E)比较RP组与对照组各注释细胞类型比例的柱状图。(F)展示9种注释细胞类型中经典标志基因表达情况的气泡图。(G)展示关键基因在各注释细胞类型中表达水平的气泡图。在(F)和(G)中,颜色越深表示表达水平越高,圆圈越大表示该细胞群中表达该基因的细胞比例越高。(A)所有细胞簇差异表达基因(differentially expressed genes,DEGs)的火山图。图中突出显示了各细胞簇中显著上调(红色)或下调(蓝色)的基因。(B)单细胞差异表达基因(single-cell differentially expressed genes,scDEGs)在细胞间表达情况的热图。橙色表示高表达,蓝色表示低表达。(C)统一流形近似与投影(Uniform Manifold Approximation and Projection,UMAP)图,展示所有细胞簇的曲线下面积(Area Under the Curve,AUC)评分。该评分用于量化前10个scDEGs基因集在每个细胞表达基因中的富集程度,颜色越浅表示富集程度越高。(D)不同注释细胞类型间AUC评分的组间比较图。
图7:
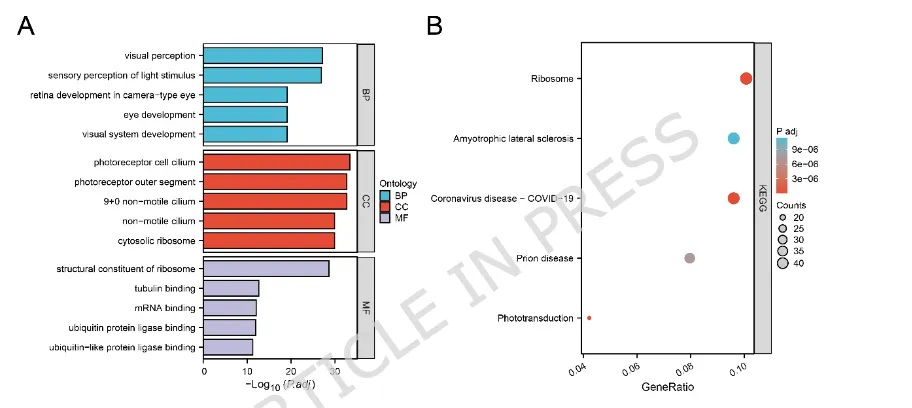
(A)差异表达基因(differentially expressed genes,DEGs)基因本体(Gene Ontology,GO)富集分析结果柱状图。富集条目分为生物过程(Biological Process,BP)、细胞组分(Cellular Component,CC)和分子功能(Molecular Function,MF)。(B)京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路富集分析结果气泡图。气泡大小表示富集基因数目,颜色表示校正后的P值(颜色越红表示校正后的P值越小)。
结论:
本研究通过整合多模态分析,建立了一个与视网膜色素变性(RP)遗传相关的程序性细胞死亡(PCD)核心基因网络。研究揭示了由免疫失调和Müller胶质细胞广泛重编程共同参与的致病网络。上述发现构建了一个连接遗传因果关系与细胞病理生理机制的系统性研究框架,为这一目前尚无根治方法的疾病提供了具有优先研究价值的机制验证靶点和潜在治疗靶点。
文章信息:
Guo N, Peng GH. Inferring genetic associations of programmed cell death genes with retinitis pigmentosa through multimodal Mendelian randomization. Mol Med. 2026.doi: 10.1186/s10020-026-01547-9.