Nat. Catal. | 郑州大学 | 碳硼烷“反套路”加成:钯催化精准构筑手性α-羧酸新路径
碳硼烷“反套路”加成:钯催化精准构筑手性α-羧酸新路径
你有没有想过,药物分子中的“手性”——就像左手和右手的关系——竟可能决定它是救命良药还是致命毒物?沙利度胺的悲剧早已警示我们:一个手性中心的错误,足以让整个分子从天使变恶魔。因此,如何高效、精准地构建手性中心,一直是化学家们梦寐以求的“圣杯”。
最近,郑州大学马艳娜研究员&陈学年教授&魏东辉教授等联合团队在《Nature Catalysis》上发表了一项突破性工作:他们首次实现了钯催化下碳硼烷对α,β-不饱和羧酸的不对称“反迈克尔型”加成,直接在羧酸的α位构建高纯度手性中心!这不仅颠覆了传统迈克尔加成的区域选择性,更为含手性α-羧酸结构的药物分子(如布洛芬、青霉素等)提供了全新合成策略。
更令人兴奋的是,这项技术巧妙地将碳硼烷(carborane)——一种含10个硼原子的笼状分子——引入有机骨架。碳硼烷可不是普通配角:它在硼中子俘获治疗(BNCT)中是理想的“硼弹头”,在药物设计中能模拟苯环却更稳定、更疏水,甚至能形成独特的二氢键靶向蛋白口袋。
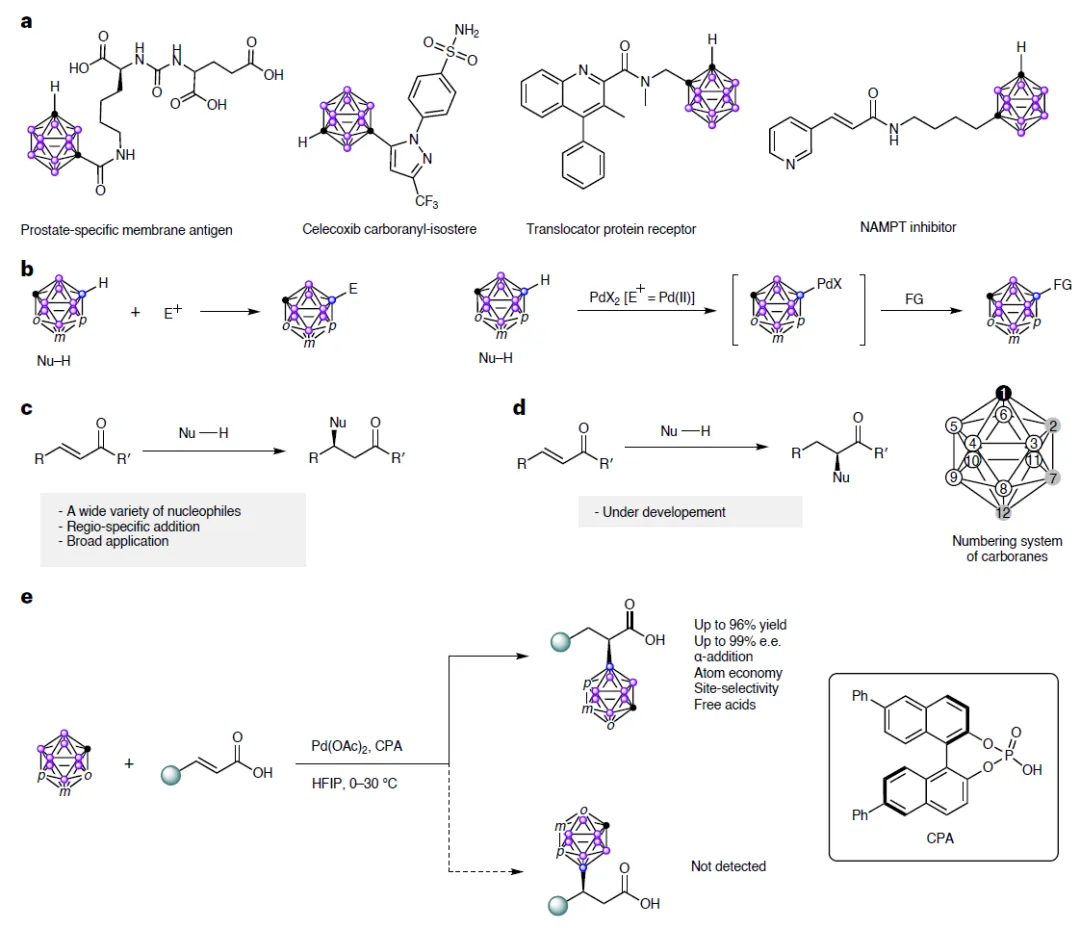
图1a:碳硼烷已成功用于前列腺特异性膜抗原抑制剂、塞来昔布类似物、转位蛋白受体配体及NAMPT抑制剂等药物设计中。
图1e:传统迈克尔加成发生在β位,而本工作通过钯催化实现α位高选择性加成
但难点在于:碳硼烷有10个几乎一模一样的B-H键,如何只让其中一个(尤其是B(9)位)精准反应?过去的方法多生成C-取代产物,而B-取代产物因B-H键极性弱、选择性差,一直难以高效获得。
研究团队另辟蹊径:他们发现,在六氟异丙醇(HFIP)溶剂中,使用醋酸钯(Pd(OAc)₂)与手性磷酸(CPA,如L11)组合,竟能实现“逆电子需求”的亲核加成——碳硼烷作为亲核试剂,进攻α,β-不饱和羧酸的α位(而非传统的β位),形成α-碳硼烷基化羧酸!
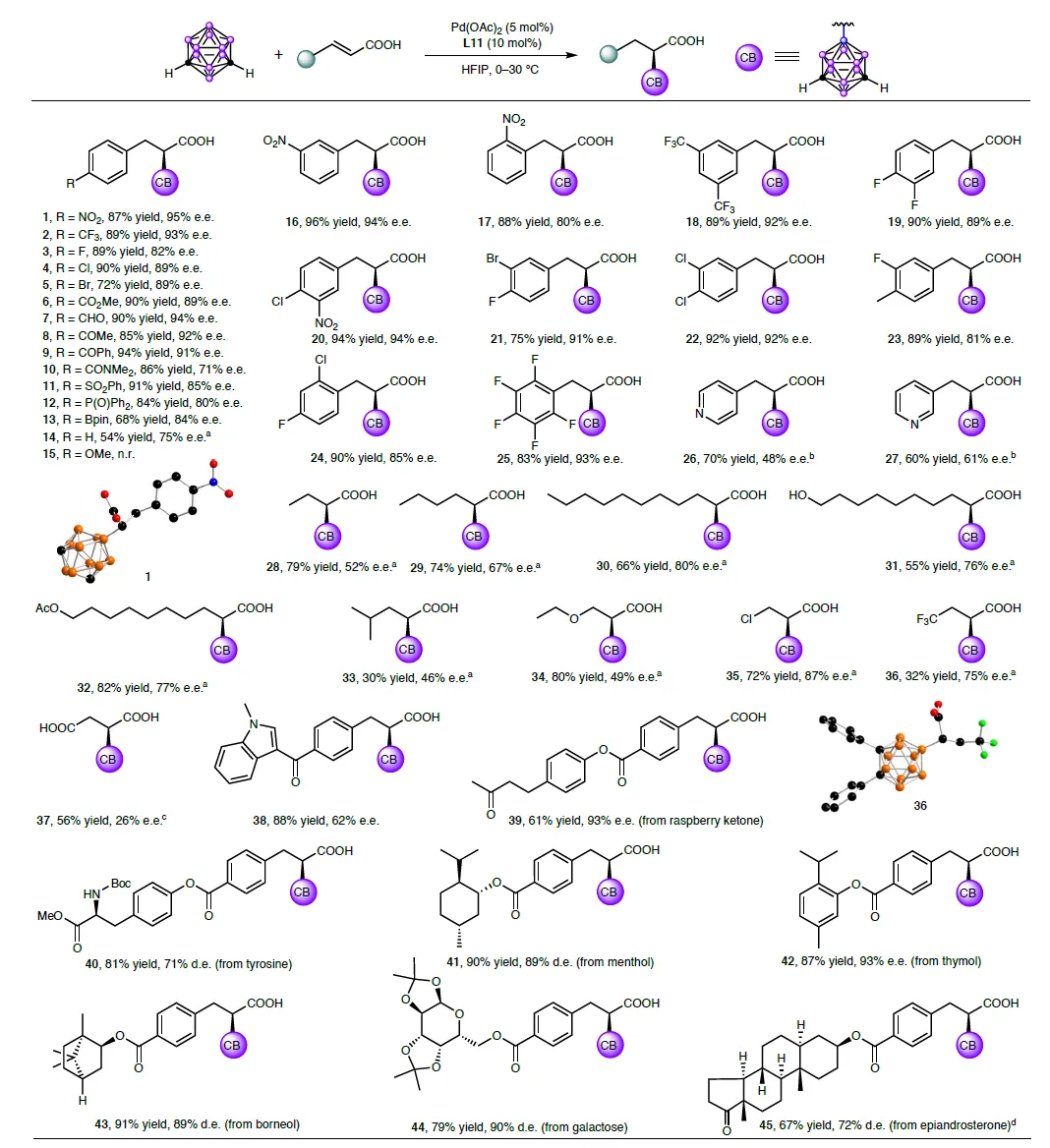
经过系统筛选,6,6'-二苯基联萘磷酸(L11)脱颖而出,以95%对映体过量(e.e.)和87%产率得到目标产物。更惊人的是,该反应对各类取代基兼容性极佳:硝基、三氟甲基、卤素、醛基、硼酸酯……甚至天然产物如覆盆子酮、薄荷醇、半乳糖衍生物,都能顺利参与反应,产率普遍在70%以上,e.e.值高达93%!
图3:包括药物分子骨架在内的多种α,β-不饱和羧酸均可高效转化
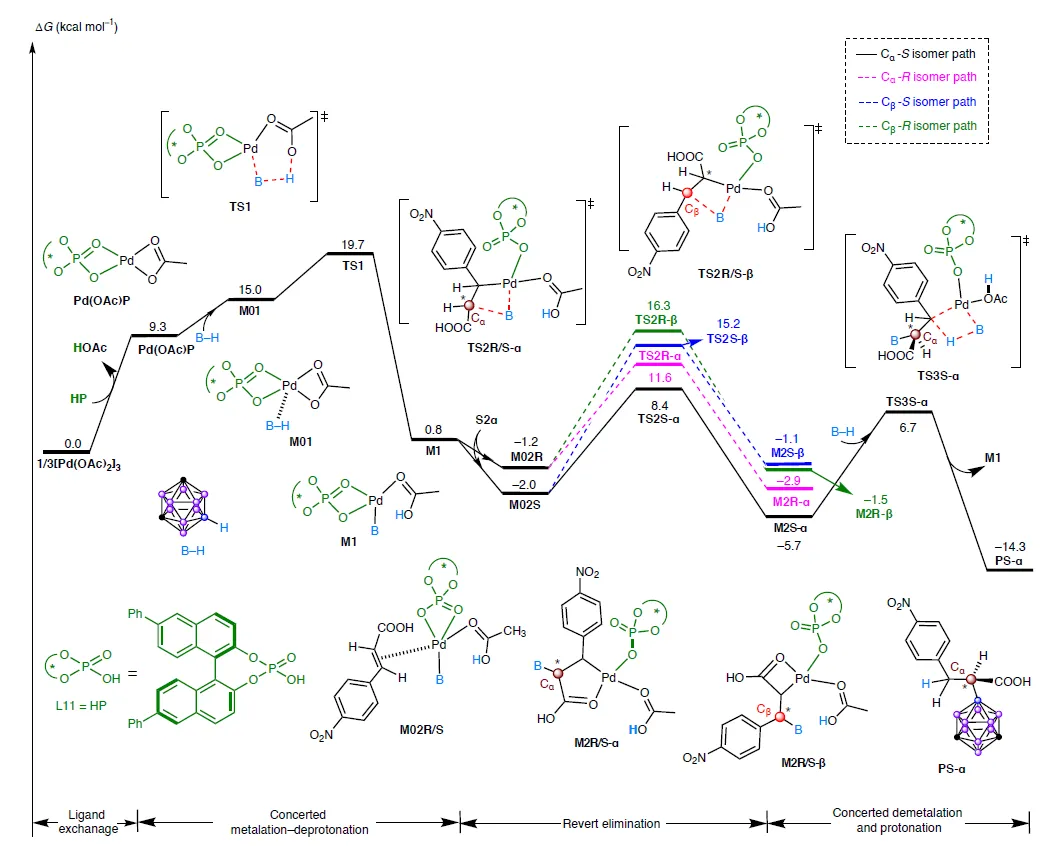
机制研究揭示了这一“反常”选择性的奥秘:
- 关键中间体反应首先通过协同金属化-去质子化(CMD)形成Pd-B键,随后烯烃插入生成一个五元环钯杂环(palladacycle)。这个刚性结构迫使加成只能发生在α位,从而实现完美的区域选择性。
- 手性来源DFT计算显示,S构型过渡态(TS2S-α)中存在更强的B-H···π和孤对电子···π等弱相互作用,使其能量比R构型低2.4 kcal/mol,完美解释了高对映选择性。
- 氢从哪来氘代实验证明:产物β位的氢原子直接来自碳硼烷的B-H键!这是一个syn-加成过程,且B-H键断裂是决速步(KIE = 3.5)。
图7:α-选择性路径(蓝线)能垒显著低于β路径(红线),且S构型过渡态更稳定
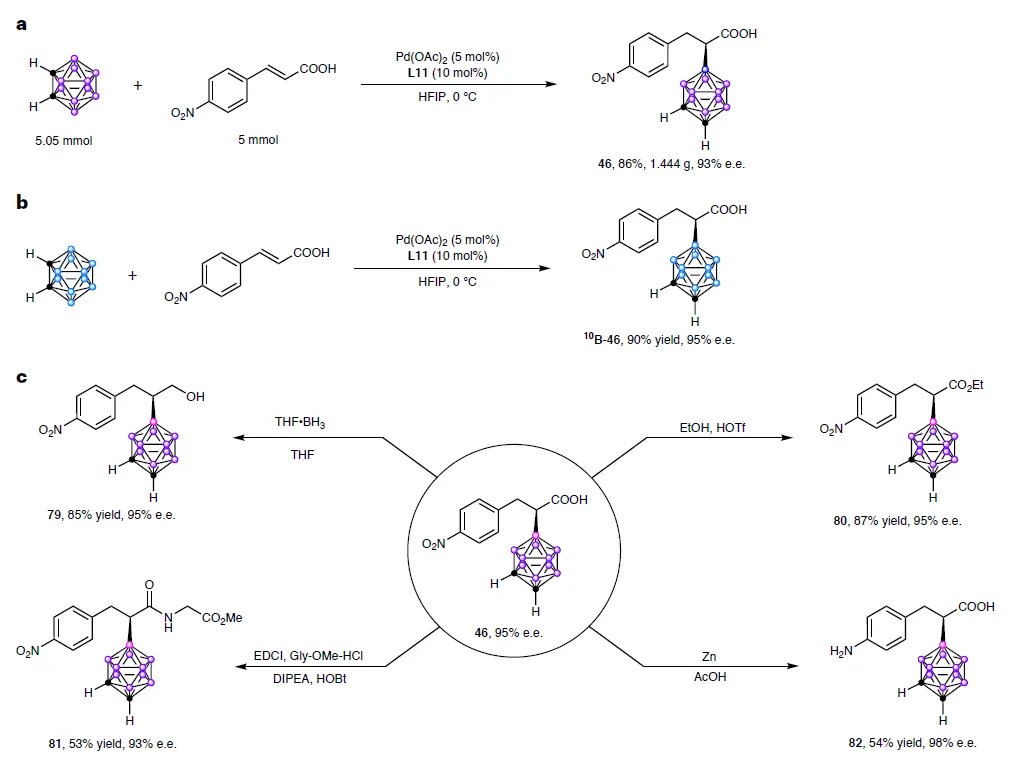
这项技术不仅适用于m-碳硼烷,o-和p-碳硼烷同样有效,甚至可使用¹⁰B富集原料制备BNCT专用药物前体。后续衍生化也极为便利:羧基可还原为醇、酯化、酰胺化,硝基可选择性还原为氨基,手性中心全程保持稳定!
图5a-b:反应可放大至克级,且适用于¹⁰B富集碳硼烷,为癌症靶向治疗提供新工具
这项工作打开了“反迈克尔型”不对称催化的大门,也为碳硼烷化学注入了新活力。未来,这类手性α-碳硼烷羧酸或许将成为新一代抗癌药、神经药物或材料单体的核心骨架。
互动时间:你觉得碳硼烷这种“超苯”结构,还能在哪些领域大放异彩?欢迎在评论区留言讨论!如果觉得这篇文章有趣,别忘了点赞、转发,让更多人看到中国科学家的前沿突破!
参考文献
Chao Lei, Wen Lu, Tingting Shen et al. Palladium-catalysed asymmetric anti-Michael-type addition of α,β-unsaturated carboxylic acids with carboranes. Nature Catalysis (2026). DOI: https://doi.org/10.1038/s41929-026-01480-4
厦门天鹭飞扬科技有限公司
电话: 13225921134
网站: www.xmtlfy.com
#碳硼烷#不对称催化#反迈克尔加成#手性羧酸#钯催化