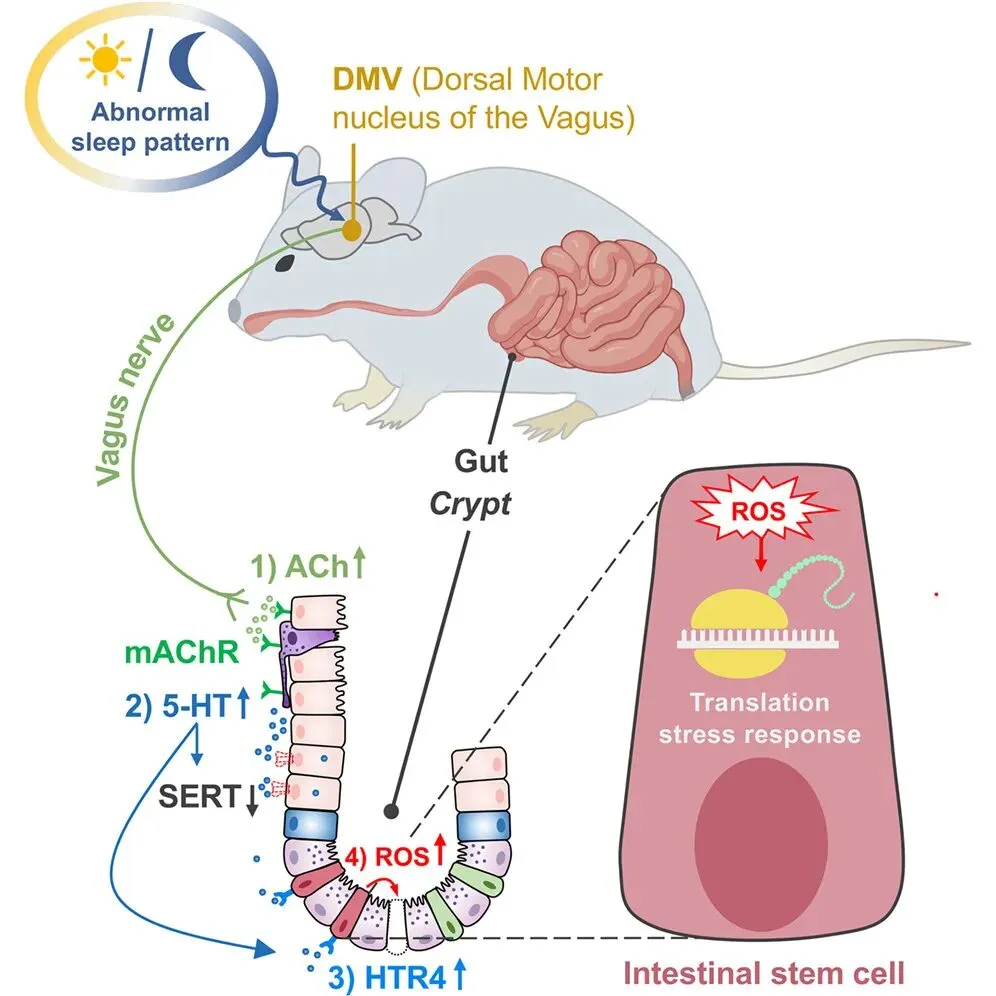
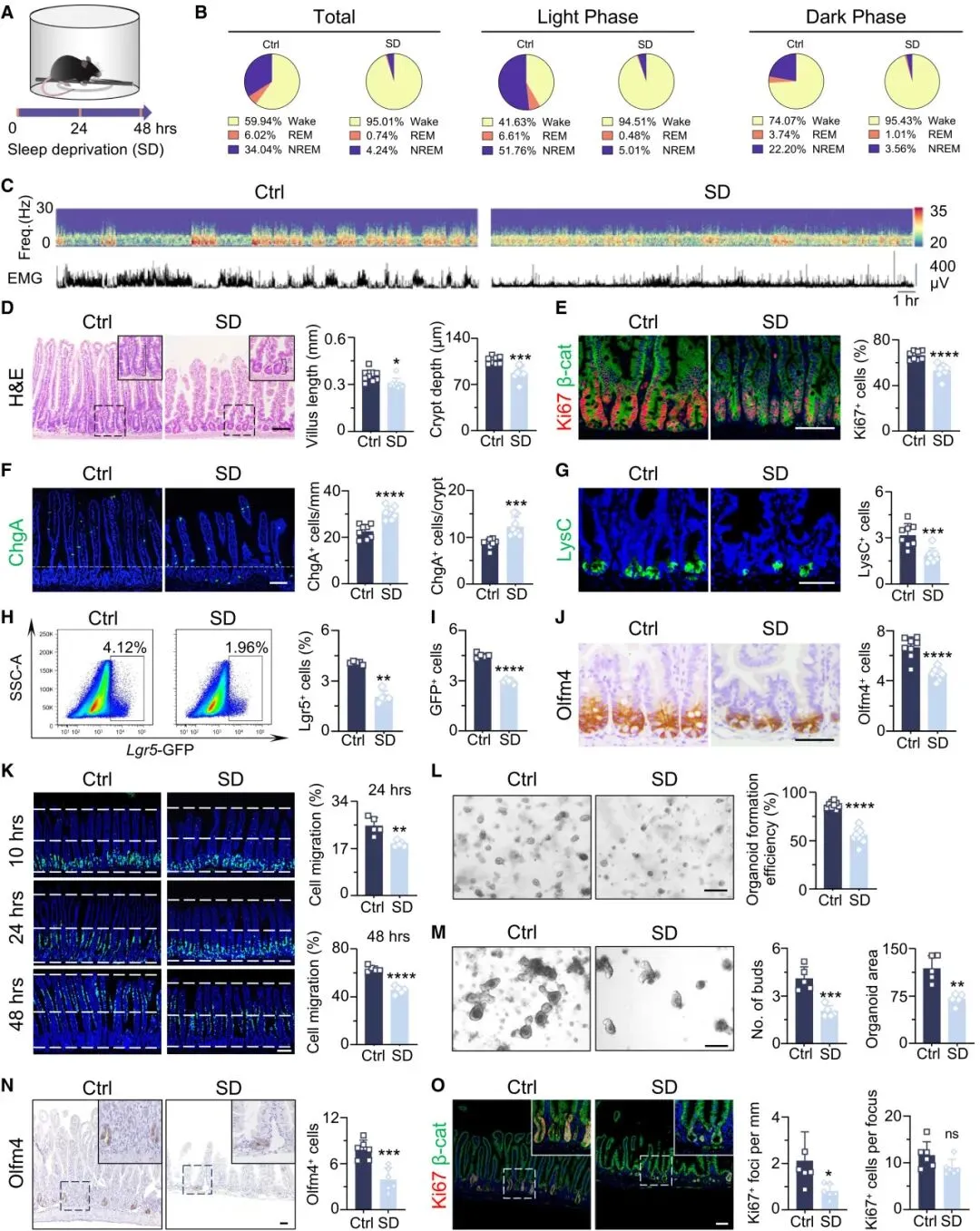
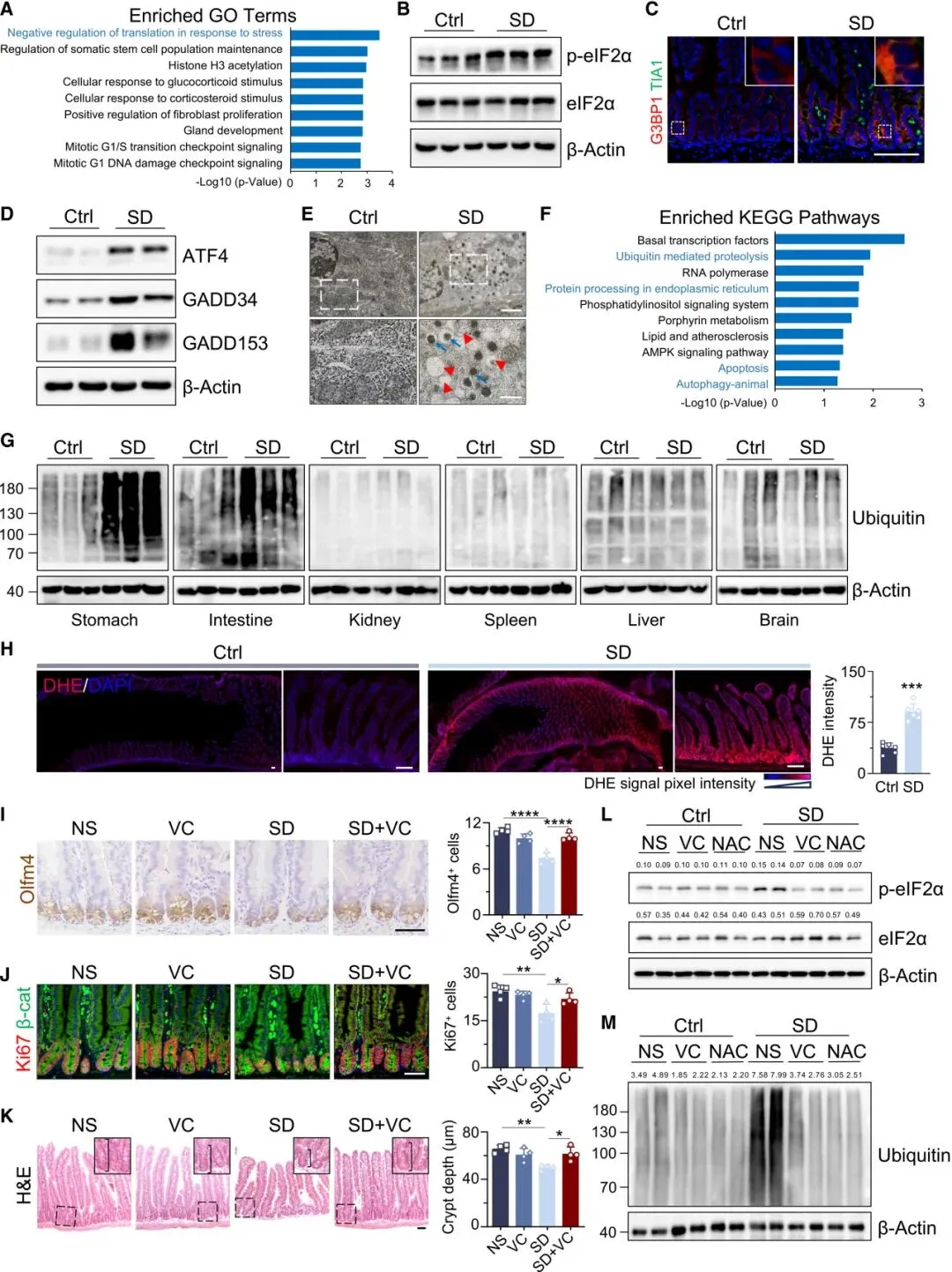
三、睡眠剥夺,扰乱肠道5-羟色胺(5-HT)稳态
靶向代谢组学和功能实验显示,SD会显著升高肠道及血清中的5-羟色胺(5-HT)水平,同时增加TrpA、5-HTP、Kyn等色氨酸代谢相关中间产物的含量。SD还会增加肠隐窝中EC细胞的数量,而EC细胞正是肠道中95%的5-HT的主要来源。
进一步研究证实,EC细胞产生的5-HT会直接引发ISC氧化应激,抑制其增殖并损害类器官生长;体内注射5-HT,可完全复制SD诱导的肠道病理表现。而Tph1基因敲除小鼠(缺乏EC细胞来源的5-HT),在SD条件下的肠道损伤和氧化应激会显著缓解。
同时,SD会通过抑制Slc6a4基因(编码血清素再摄取转运蛋白SERT)的表达及启动子甲基化,降低5-HT的再摄取能力,从而维持肠道局部5-HT的高浓度——这意味着,SD诱导的5-HT激增,既是EC细胞分泌增加的结果,也是再摄取减少的结果,是介导SD相关肠道病理的关键。
四、HTR4受体,介导5-HT对ISC的损伤效应
单细胞RNA测序结果显示,肠道干细胞(ISC)中主要表达5-HT受体HTR4,且急性SD后,HTR4在肠隐窝中的转录本和蛋白质水平会显著上调。
体外实验证实:HTR4激动剂prucalopride可模拟5-HT的作用,抑制类器官生长、增加ROS和泛素化蛋白水平;而HTR4拮抗剂SB-203186则可逆转这些损伤效应。体内实验结果一致:HTR4激动剂会诱发SD样肠道病理,而HTR4拮抗剂GR113808则能缓解SD引起的ISC减少、增殖下降、潘氏细胞丢失及氧化应激升高。
此外,肠道上皮特异性Htr4条件性敲除小鼠(Htr4 cKO),在SD条件下会表现出明显的保护作用,其衍生的类器官对5-HT刺激后的ROS水平显著降低——这进一步证实,ISC对过量5-HT的病理反应,是通过上皮细胞上的HTR4受体介导的。
五、乙酰胆碱(Ach),连接脑肠信号的关键介质
研究发现,SD小鼠的肠道乙酰胆碱(Ach)水平显著升高;腹腔注射Ach或其稳定类似物carbachol,可完全复制SD样肠道表型,包括隐窝-绒毛缩短、ISC和增殖细胞及潘氏细胞减少、氧化应激增加,以及HTR4和5-HT水平升高。
机制层面,这一效应主要通过肠道上皮的毒蕈碱受体Chrm3介导:Chrm3条件性敲除小鼠在SD条件下,肠道5-HT水平不会升高,肠道病理也得到明显保护。体外实验进一步证实,Ach会直接促进EC细胞释放5-HT,并抑制5-HT再摄取载体SERT的功能,从而维持肠道5-HT的高浓度,最终引发肠道病理。
六、DMV-迷走神经通路,是脑肠信号传递的核心
睡眠信号的调控源于中枢神经系统,研究团队通过c-Fos免疫荧光染色和伪狂犬病毒(PRV-724)逆行追踪发现:SD会激活多个中枢神经核团,但只有迷走神经背核(DMV),能通过迷走神经(VN)将睡眠缺失信号传递至肠道。
SD会导致DMV神经元的钙信号频率和幅度显著升高,且这种异常激活会持续长达7天;化学激活DMV神经元,会导致肠道Ach和5-HT水平显著升高,模拟SD效应;而化学抑制DMV神经元,则能有效逆转SD诱导的肠道Ach、5-HT激增及ISC减少。
此外,双侧迷走神经切开术(VGX)可显著减轻SD诱导的肠道损伤,使5-HT水平恢复正常,进一步证实迷走神经是DMV向肠道传递信号的关键载体。