心力衰竭(HF)源于高血压、瓣膜病、心肌梗死或遗传因素导致的慢性心脏压力或损伤。在病程早期,成熟心肌细胞通过增大体积形成代偿性肥厚以维持心输出量;但持续的超负荷最终会引发病理性肥厚与功能失代偿,导致心脏衰竭。研究表明,基因转录、钙稳态、代谢、氧化应激及炎症等多种机制共同驱动了这一过程。深入解析心肌肥厚的发病机制,对于开发阻断肥厚向心衰进展的精准治疗方案具有重要意义。2026年1月,郑州大学附属第一医院王小芳团队在《Circulation Research》发表题为“OTUD7a Accelerates Pathological Cardiac Hypertrophy via TAK1 Activation”的研究论文 。该研究揭示了去泛素化酶OTUD7a通过抑制TAK1的泛素化降解,从而增强其稳定性并激活下游信号通路,最终加剧病理性心肌肥厚。
1. OTUD7a在肥厚心肌组织及心肌细胞中显著上调
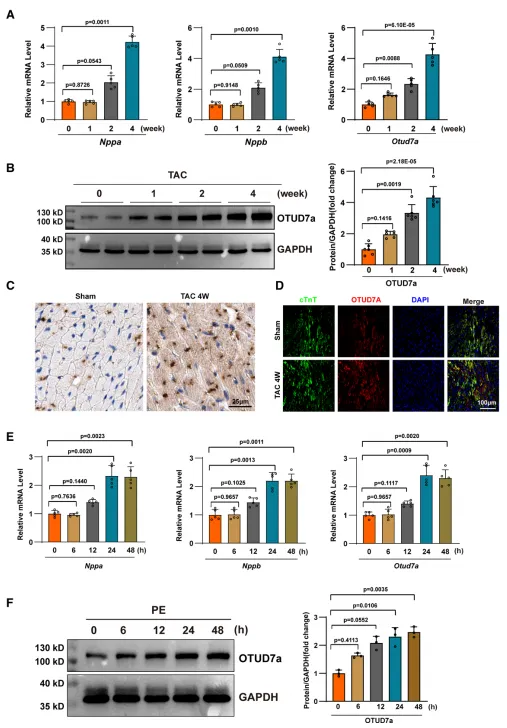
为了探讨OTUD7a与心肌肥厚的关系,研究团队在多种模型中检测了其表达水平 。动物模型:在主动脉弓缩窄术(TAC)诱导的小鼠压力负荷心肌肥厚模型中,心脏组织内OTUD7a的mRNA和蛋白水平在术后1周显著升高,并随时间持续增加 。细胞模型:在体外利用苯肾上腺素(PE)处理新生大鼠心肌细胞(NRCMs),发现OTUD7a的mRNA和蛋白表达在PE诱导24和48小时后翻倍。
2. OTUD7a缺失可缓解压力负荷诱导的心肌功能损伤
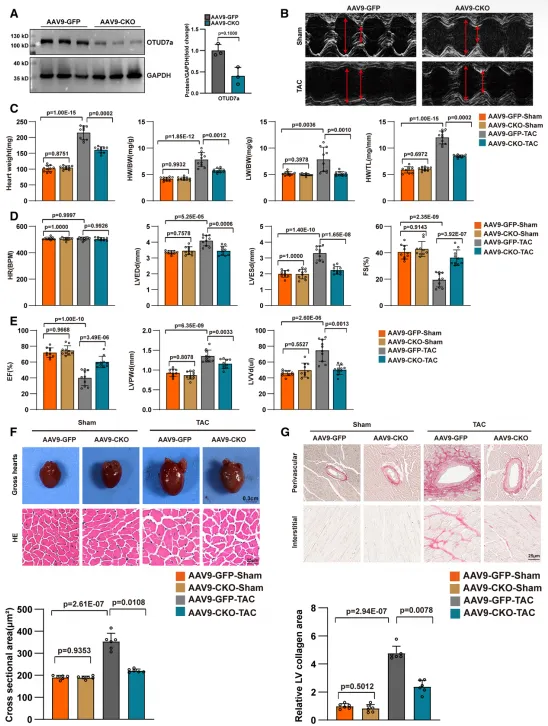
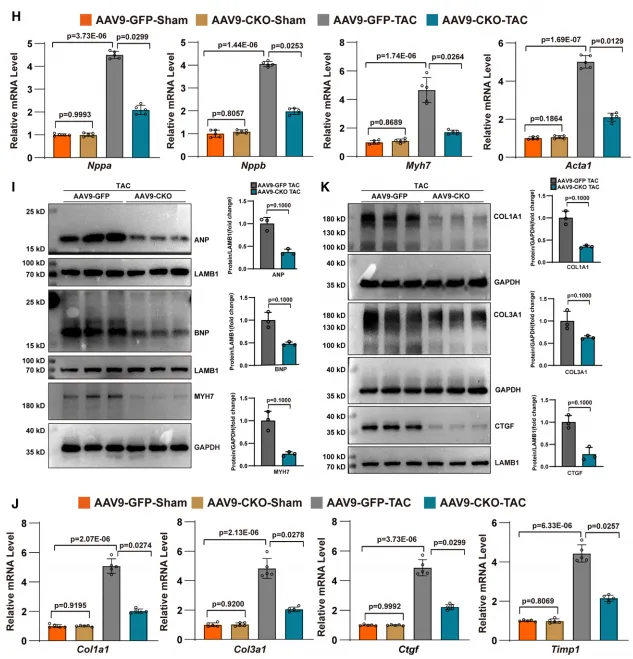
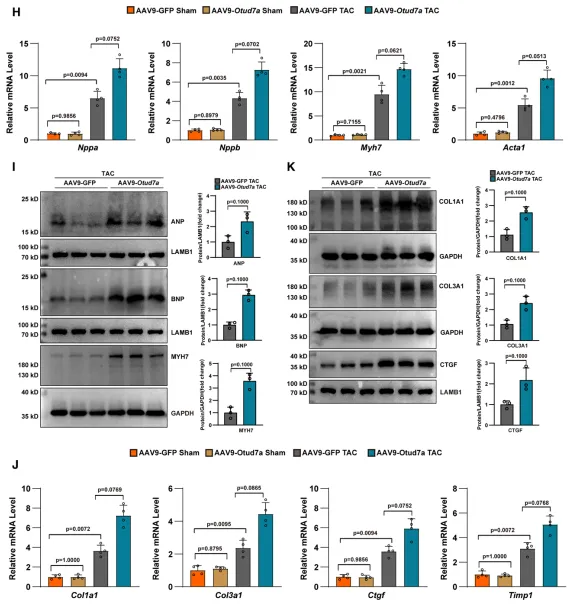
为探究 OTUD7a 在体内的功能,研究团队通过构建心肌特异性敲除(CKO)小鼠并建立 TAC 诱导的心肌肥厚模型发现,OTUD7a 的缺失展现出显著的心脏保护效应。实验结果显示,OTUD7a 缺失有效逆转了压力负荷导致的射血分数(LVEF)与短轴缩短率(FS)下降,维持了正常的左室腔大小与壁厚,显著改善了心功能。在结构重塑方面,OTUD7a 缺失不仅降低了心脏重量比(HW/BW、HW/TL)及肺水肿程度(LW/BW),还显著减小了心肌细胞横截面积并减轻了间质纤维化;同时在分子水平上,肥厚标志基因(Nppa、Nppb、Myh7)及纤维化指标(Col1a1、Ctgf)的异常上调也得到了明显抑制。综上表明,OTUD7a 缺失能有效抵御压力负荷引发的心肌肥厚与功能失代偿,且该保护作用具有应激依赖性,在基础生理状态下并不产生影响。3. 心脏特异性过表达OTUD7a加剧心脏重塑
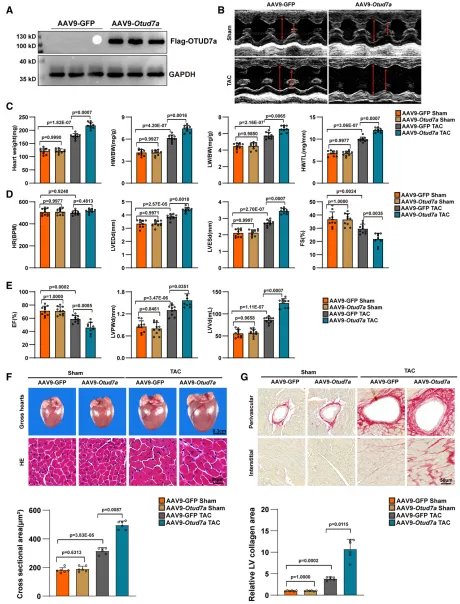
为进一步验证 OTUD7a 的功能,研究团队利用 AAV9 系统在小鼠心脏中特异性过表达 OTUD7a。结果显示,在 TAC 压力负荷应激下,过表达 OTUD7a 会显著加剧表型恶化,使小鼠表现出更严重的心功能障碍和更明显的心脏扩大;组织学分析等病理表现进一步证实,过表达组的心肌细胞体积显著增大,并伴随更为严重的间质纤维化和胶原沉积,表明 OTUD7a 在病理性心肌肥厚进程中具有明确的促进作用。
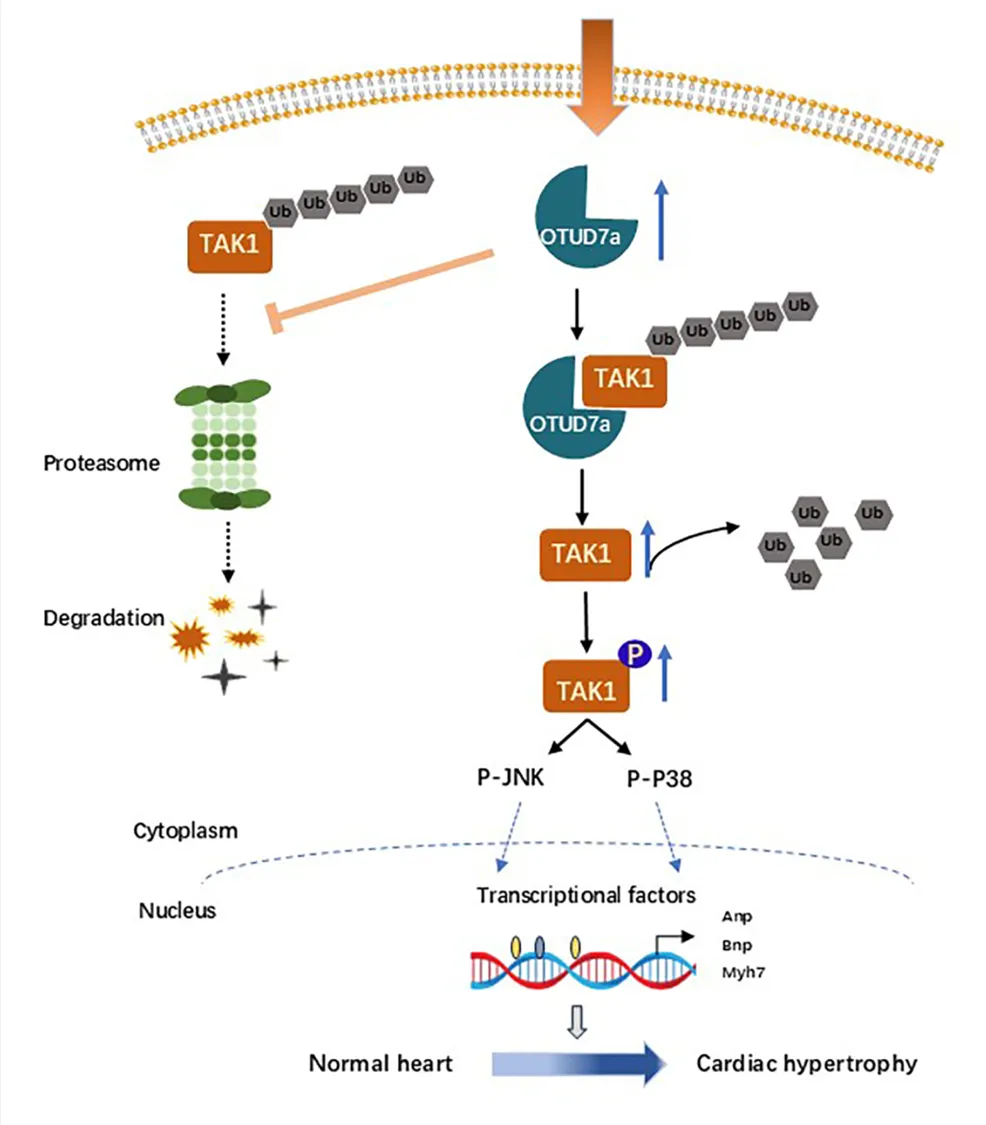
4. OTUD7a通过激活TAK1-JNK/p38通路发挥促肥厚作用
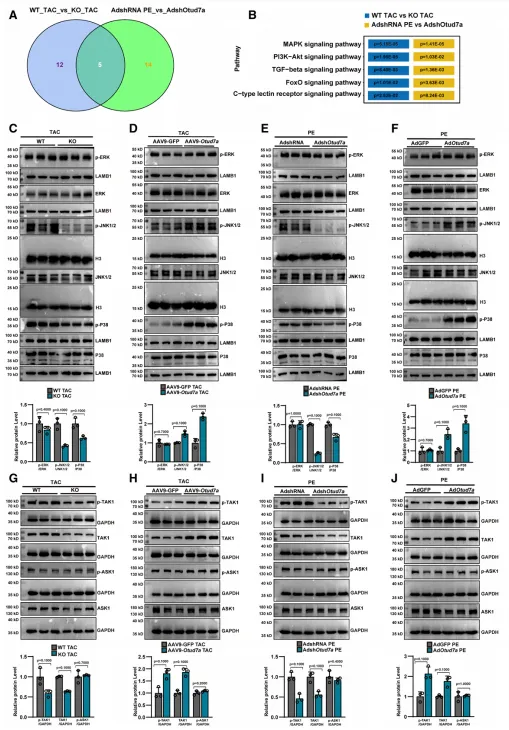
通过对敲除组与野生型组小鼠心脏及细胞进行 KEGG 通路分析,研究确认 MAPK 信号通路是受 OTUD7a 影响最显著的下游通路。在 TAC 或 PE 诱导的心肌肥厚模型中,OTUD7a 的过表达会显著上调 TAK1 的总水平及其磷酸化(p-TAK1)水平,进而激活下游的 p-JNK 和 p-p38 信号轴,且该活化过程表现出明确的时间依赖性;相反,敲除或敲低 OTUD7a 则能有效抑制该轴线的活化。此外,这种调控机制具有应激依赖性,在基础生理状态下 OTUD7a 虽能影响 TAK1 的基础表达,但不足以诱发其磷酸化活化。综上所述,OTUD7a 主要通过稳定并激活 TAK1-JNK-p38 信号轴,从分子水平驱动并加剧了病理性心肌肥厚的恶化进程。
5. OTUD7a 通过调控 TAK1 活性介导心肌肥厚进程
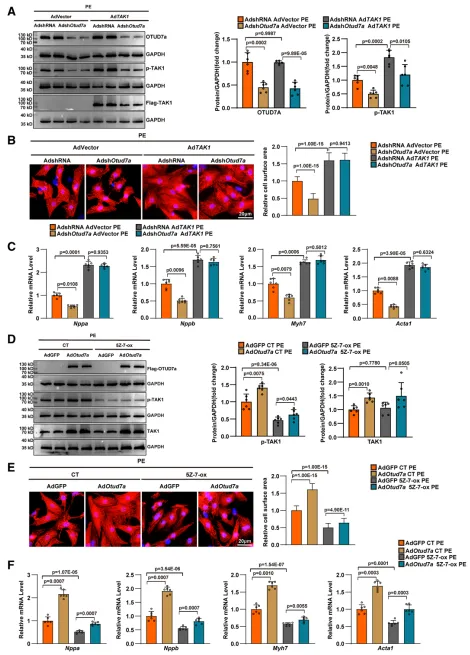
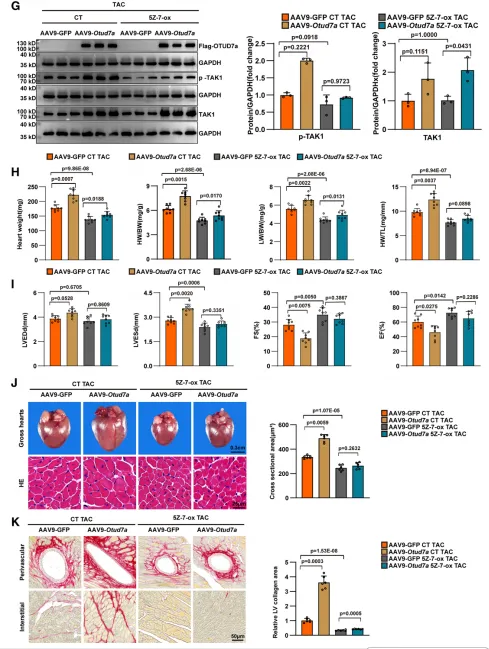
研究通过体内外“挽救实验”明确了TAK1 在OTUD7a 促肥厚功能中的核心地位。在体外模型中,过表达TAK1 能够完全抵消OTUD7a 缺失带来的保护效应;而在体内外 OTUD7a 过表达模型中,使用TAK1 特异性抑制剂5Z-7-Ox 可显著抑制TAK1 的磷酸化激活,从而逆转 OTUD7a 导致的心功能恶化、心肌细胞肥大及间质纤维化。以上结果充分证实,OTUD7a 对病理性心肌肥厚的促进作用严格依赖于其对下游TAK1 信号轴的激活。6.OTUD7a 与 TAK1 直接结合促进心肌肥厚
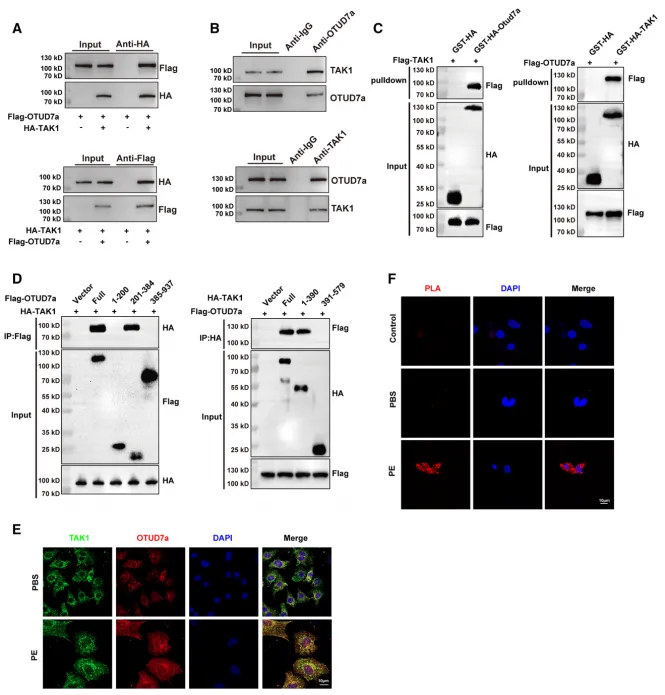
为了明确这些靶蛋白之间的关系,研究人员通过免疫共沉淀(Co-IP)、GST pull-down 以及邻位连接技术(PLA)等多种手段,证实了 OTUD7a 与 TAK1 在心肌细胞质中存在直接的物理相互作用,并明确了二者结合的核心区域分别为 OTUD7a 的 201-384 位氨基酸结构域和 TAK1 的 1-390 位氨基酸结构域。
7.OTUD7a 通过去泛素化作用稳定TAK1 蛋白
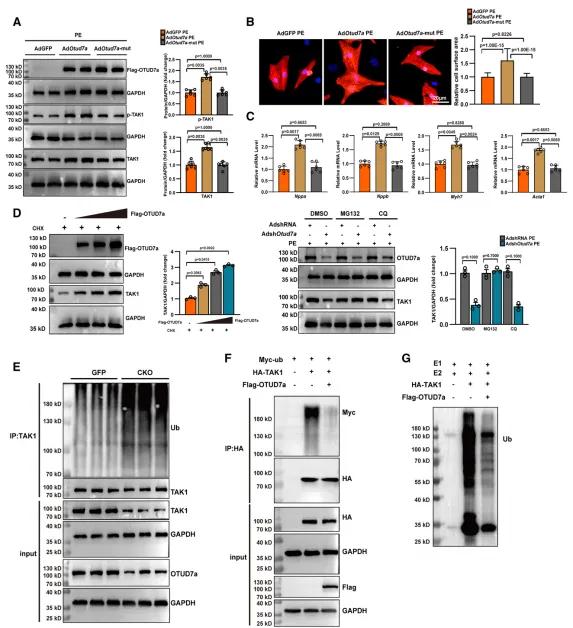
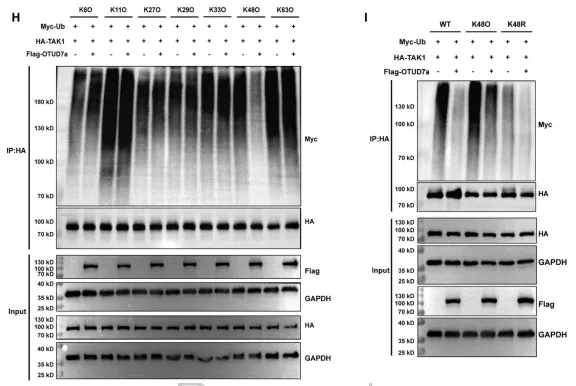
研究证实,OTUD7a 是一种关键的去泛素化酶,通过其酶活性维持 TAK1 蛋白的稳定性。具体而言,OTUD7a 以浓度依赖的方式上调 TAK1 水平,其核心机制是特异性移除 TAK1 上的 K48 链接泛素链,从而阻断泛素-蛋白酶体途径介导的蛋白降解。实验表明,当 OTUD7a 发生突变失去去泛素化功能,或被蛋白酶体抑制剂 MG132 干预时,这种调控效应会随之改变,证明了 OTUD7a 稳定 TAK1 是驱动心肌肥厚病理性恶化的核心分子环节。原文链接:https://doi.org/10.1161/CIRCRESAHA.125.326647
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
