论文概览
钙钛矿/有机叠层太阳电池(TSC)凭借其溶液加工性和高效的光谱互补利用,成为突破单结Shockley-Queisser极限的重要路径。然而,宽带隙(WBG,E_g > 1.8 eV)钙钛矿子电池中严重的卤素相分离和离子迁移导致显著的电压损失,制约了器件性能。针对这一关键挑战,郑州大学张懿强团队联合香港理工大学、中国科学院化学研究所等机构,创新性提出缺电子胺分子设计策略。通过在二氨基芴骨架中引入强吸电子砜基(-SO₂),成功将传统胺基从“阳离子配位碱”重构为“卤素靶向氢键供体”,实现了对溴离子的定向N–H⋯Br⁻氢键锚定。该设计稳定了富溴中间相(DMSO-PbBr₂/DTD配合物),抑制了溴的过早成核,促进了钙钛矿薄膜中卤素的均匀垂直与水平分布,同时提高了卤素离子迁移的活化能垒。最终,1.85 eV宽带隙钙钛矿太阳电池(PSC)实现了19.32%的认证效率,并在连续光照下展现出优异的工作稳定性。集成至钙钛矿/有机叠层器件后,获得了26.76%的冠军效率和2.216 V的开路电压,为已报道的E_g > 1.8 eV体系中的最高值之一。该研究以"Electron‐Deficient Amines Enable Halide‐Anchoring Hydrogen Bonding for Stable Wide‐Bandgap Perovskites Toward Perovskite/Organic Tandem Solar Cells"为题发表在顶级期刊Angewandte Chemie International Edition上。
技术亮点
缺电子胺分子设计:通过在二氨基芴(DAF)中引入强吸电子砜基(-SO₂),获得二氨基二苯并噻吩砜(DTD),显著降低氮原子电子云密度,增强N–H键极性,将其转化为高效氢键供体。
卤素选择性锚定:DTD优先与Br⁻形成强N–H⋯Br⁻氢键(较I⁻更强),选择性稳定DMSO-PbBr₂中间体,延缓富溴相过早成核,实现卤素均匀合金化。
抑制离子迁移:DTD中的-SO₂与Pb²⁺配位,同时-NH₂与卤素氢键键合,双重锚定作用提升卤离子迁移活化能垒,有效抑制光/电诱导的相分离。
优异器件性能:单结WBG PSC(1.85 eV)PCE达19.32%,VOC高达1.377 V,FF为83.81%;叠层器件PCE达26.76%,VOC为2.216 V,T90寿命超过900小时。
研究意义
✅突破宽带隙钙钛矿电压瓶颈:通过分子电子结构调控,将胺基功能从“与FA⁺作用”转变为“与Br⁻锚定”,有效抑制卤素相分离,大幅提升VOC。
✅建立分子结构-功能新范式:揭示了吸电子基团修饰对胺基氢键能力的调控机制,为功能添加剂设计提供了通用指导原则。
✅实现结晶动力学精准调控:通过选择性稳定富溴中间体,同步优化卤素垂直/水平分布,提升薄膜质量与均匀性。
✅显著增强工作稳定性:抑制离子迁移和相分离使器件在MPP跟踪下T90超过900小时,为宽带隙钙钛矿商业化奠定基础。
深度精读
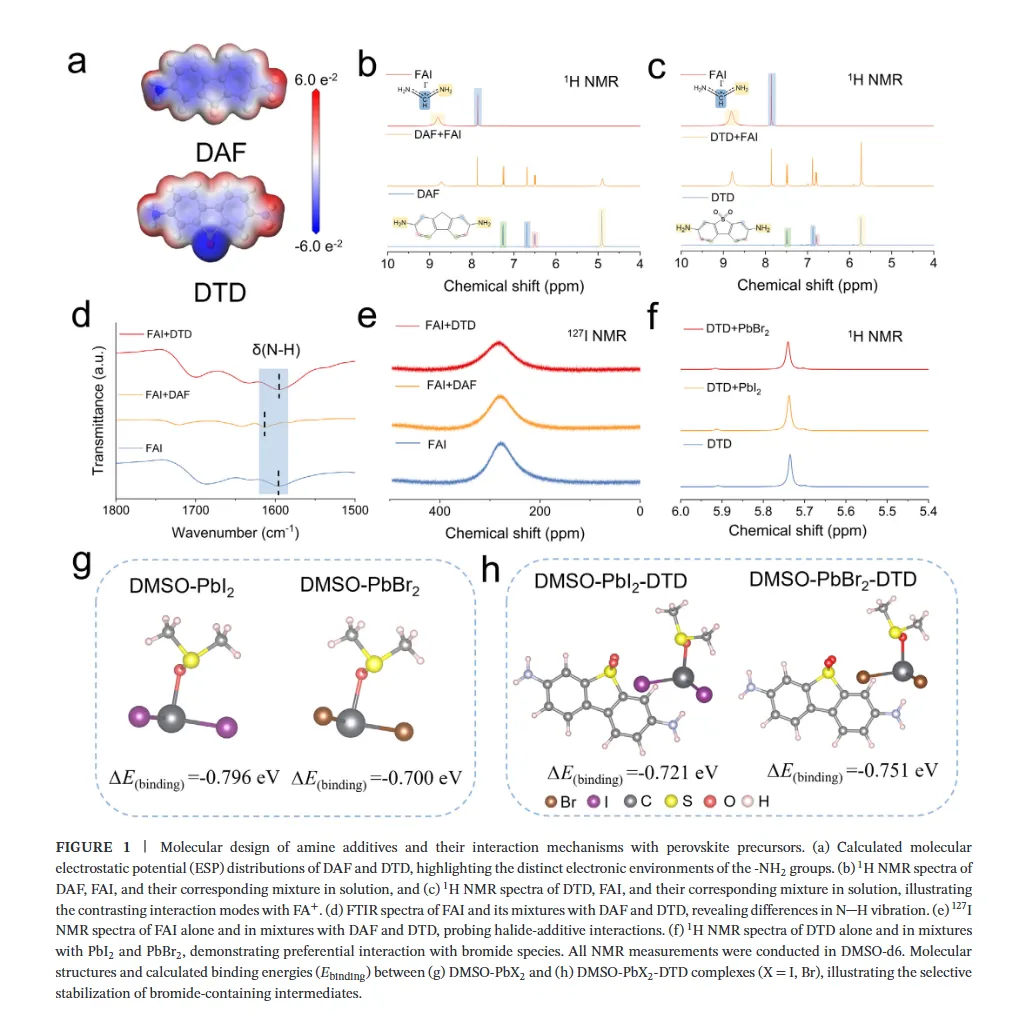
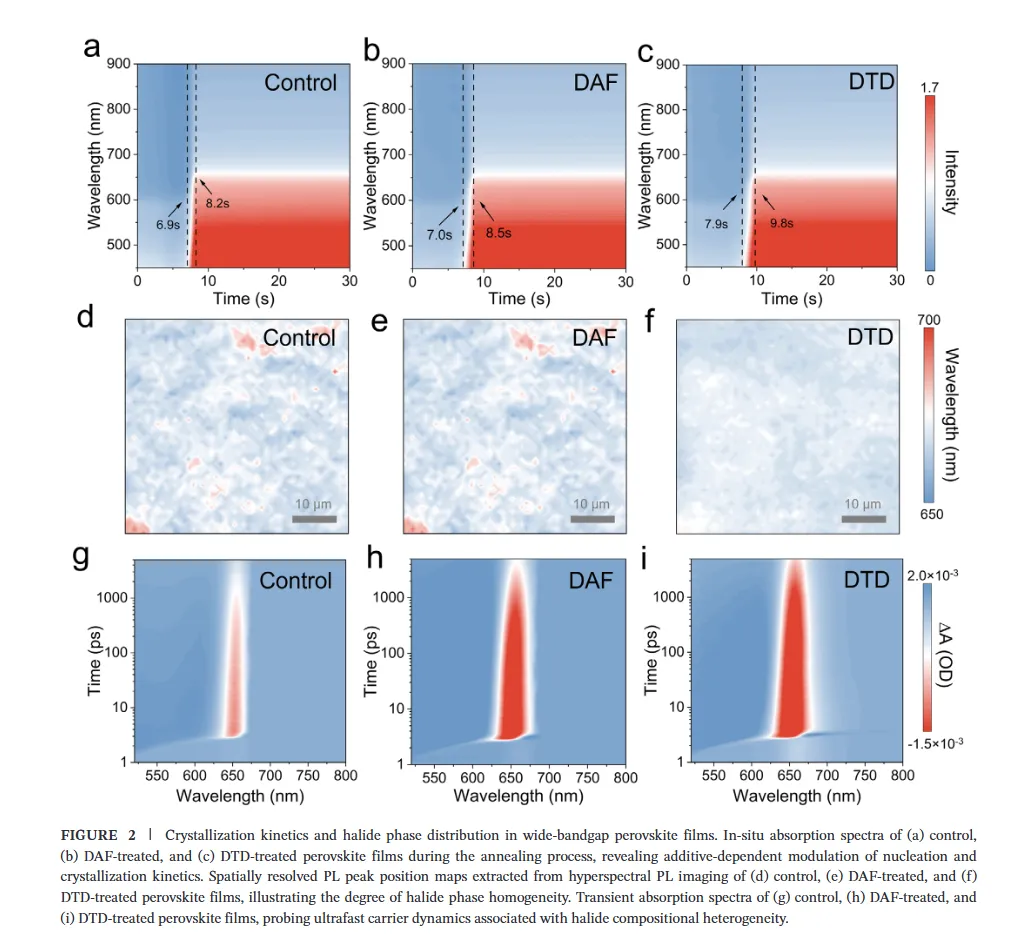
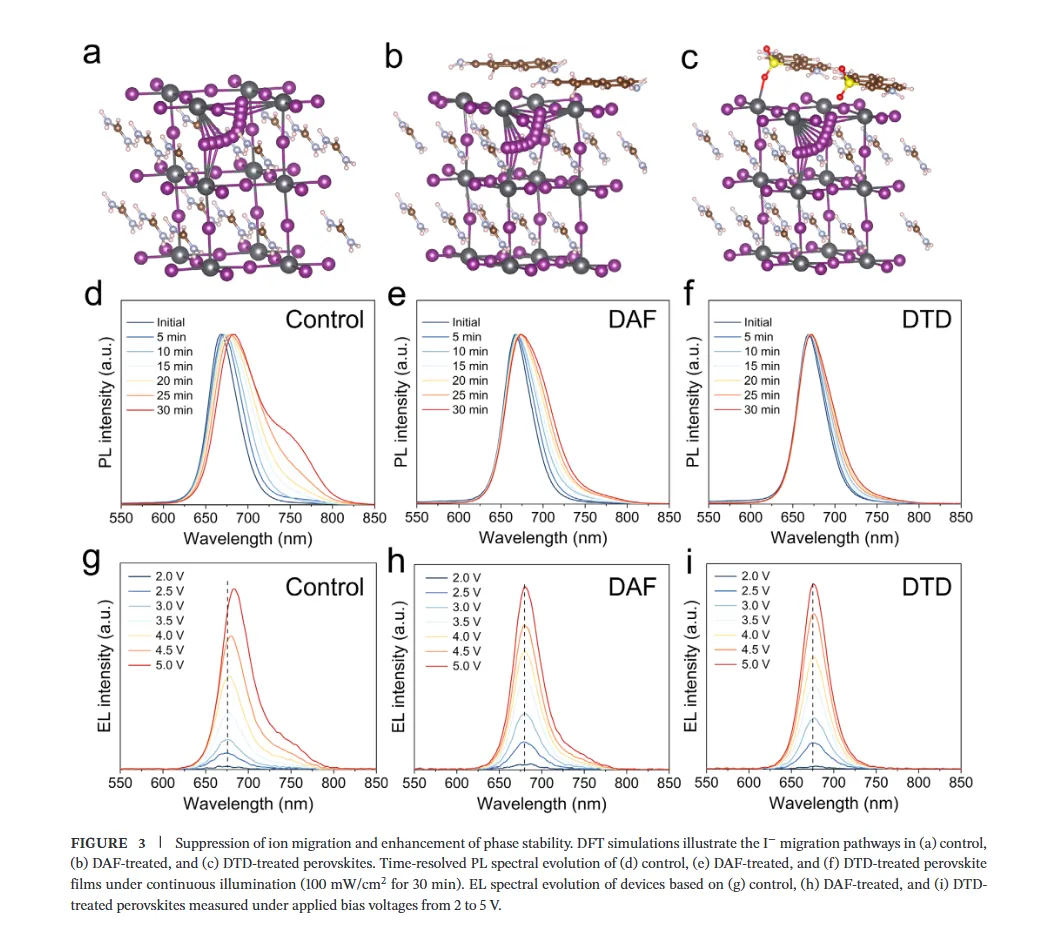
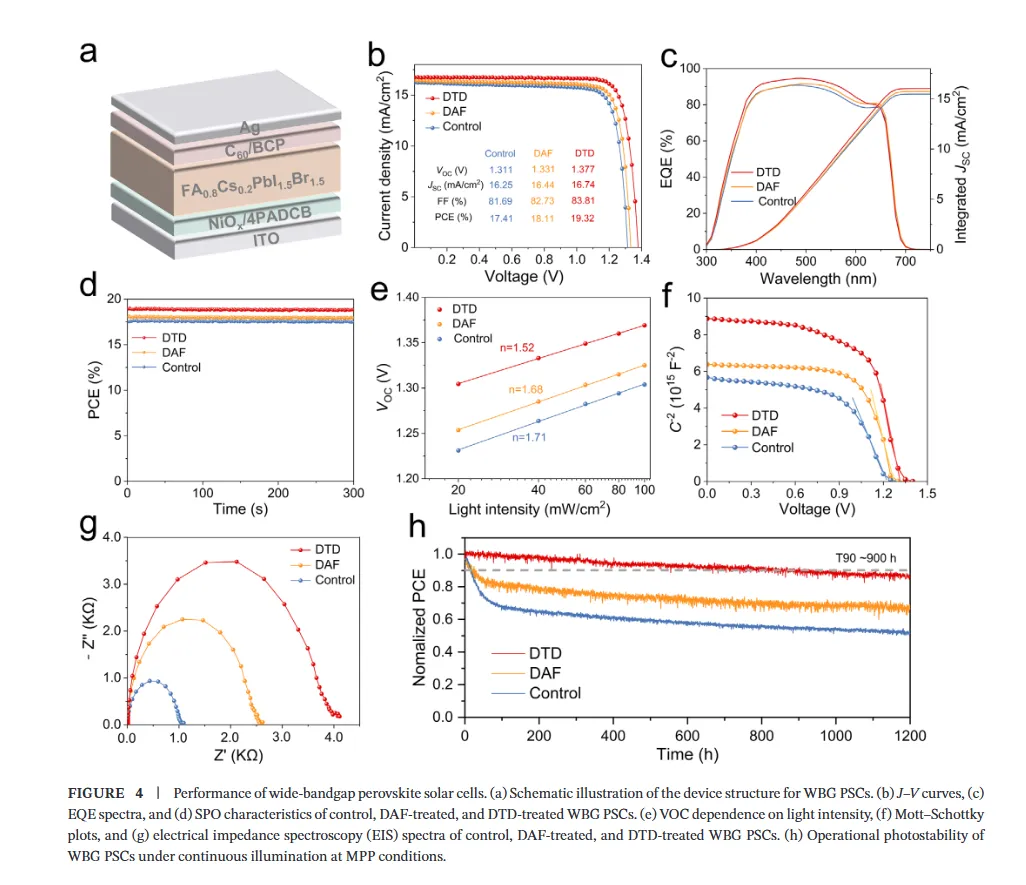
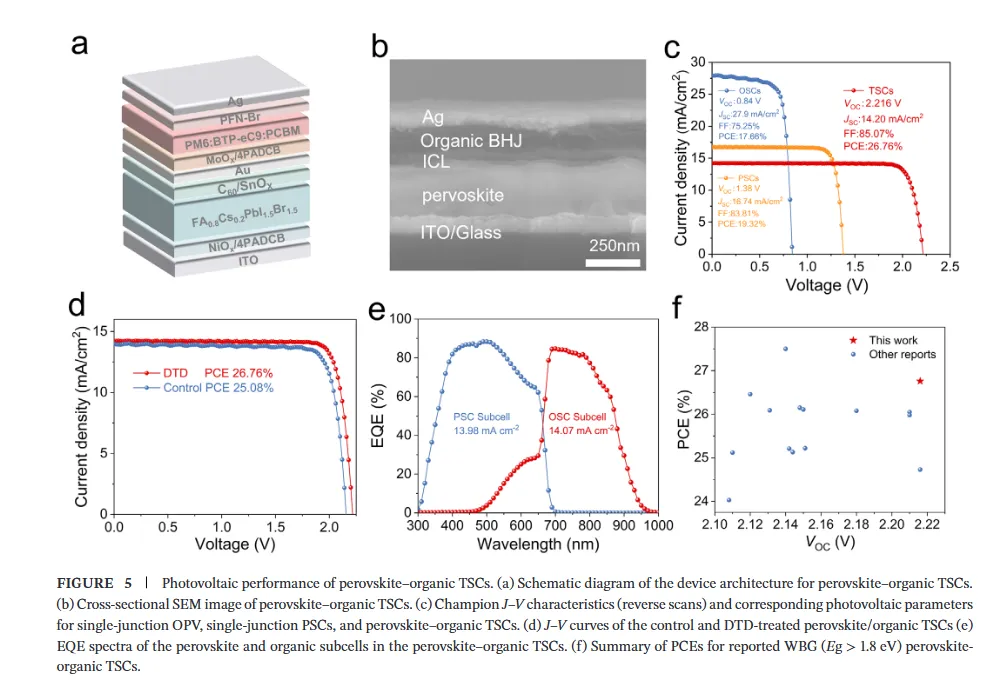
图1(分子设计与相互作用机制):该图展示了二氨基芴(DAF)与二氨基二苯并噻吩砜(DTD)的分子设计策略及相互作用机理,静电势分布图揭示了DTD中强吸电子的砜基(-SO2)如何显著降低氨基氮原子的电子密度并增强N-H键的极性,从而将-NH2官能团从富电子胺重构为高效的卤化物靶向氢键供体;核磁与红外光谱数据进一步证实DTD与卤素离子(尤其是Br-)形成了稳健的N-H···Br-氢键,而DFT计算则表明DTD与DMSO-PbBr2中间体的结合能更低,这种选择性稳定作用能有效延缓溴化物成核,为调控宽禁带钙钛矿的结晶动力学提供了分子层面的理论基础。图2(结晶动力学与卤化物分布):该图通过原位吸收光谱监测了薄膜退火过程中的结晶动力学,结果显示DTD处理的钙钛矿薄膜表现出显著延长的成核阶段,表明其通过强氢键作用限制了Br-的迁移率并抑制了过早的富Br成核;光致发光(PL)Mapping和瞬态吸收(TA)光谱结果表明,DTD处理的薄膜在空间上表现出均匀的PL发射且GSB峰红移极小(仅1.10 nm),证明了其能有效抑制横向和纵向的卤化物相分离,实现了体相与埋底界面处均匀的卤化物分布,解决了传统宽禁带钙钛矿中因I/Br结晶动力学差异导致的组分梯度问题。图3(离子迁移抑制与稳定性):该图利用DFT计算揭示了碘离子在晶格内的迁移路径及能垒,证明DTD处理显著提高了卤化物离子迁移的活化能,这归因于-SO2基团与Pb2+配位以及-NH2与卤素形成双向锚定作用,从而加固了晶格结构;光致发光(PL)随时间演化和电致发光(EL)在不同偏压下的测试结果显示,DTD处理的器件在持续光照和高电压偏压下均未出现明显的峰位红移,而对照样则表现出严重的相分离,结合SCLC测得的更低陷阱密度,确凿地证明了DTD在抑制离子迁移、提升光稳定性和电学稳定性方面的卓越效能。图4(单结器件性能与物理机制):该图展示了基于倒置结构的1.85 eV宽禁带钙钛矿太阳能电池的光伏性能,冠军器件实现了19.32%的功率转换效率(PCE),并具有高达1.377 V的开路电压和83.81%的填充因子,显著优于对照组;通过开路电压与光强关系、Mott-Schottky分析、电化学阻抗谱(EIS)及稳态功率输出测试,作者阐明了DTD通过减少缺陷辅助的非辐射复合、增强内置电场以及提高界面复合电阻来优化电荷提取的机制,使得器件在最大功率点追踪下表现出超长的运行稳定性(T90 > 900小时)。图5(叠层器件集成与性能):该图展示了钙钛矿/有机串联太阳能电池的结构设计与光伏性能,得益于前子电池优异的相稳定性和电压输出,所制备的串联器件实现了26.76%的冠军效率和2.216 V的高开路电压,处于同类宽带隙顶电池串联器件的最高水平;EQE光谱分析证实了前后子电池之间完美的电流匹配(积分电流均约14 mA/cm²),且DTD浓度优化实验表明适量添加剂能最大化界面质量,这项工作成功地将分子尺度的卤化物化学调控转化为宏观器件的高效与稳定输出,为下一代串联光伏提供了通用策略。结论展望
本研究通过缺电子胺(DTD)的分子设计,成功将传统胺基从“阳离子配体”转变为“卤素锚定氢键供体”,实现了对宽带隙钙钛矿中溴离子的选择性稳定和结晶动力学的精准调控。该策略不仅抑制了光/电诱导的卤素相分离和离子迁移,还大幅降低了缺陷密度和非辐射复合,使得1.85 eV单结器件效率突破19.3%,叠层器件效率达26.76%,且工作稳定性(T90 > 900 h)远超同类体系。这项工作建立了一个通用的分子结构-功能关系范式,为开发高效、稳定、高电压的钙钛矿基叠层光电器件提供了可推广的路径。展望未来,通过进一步优化分子骨架(如调控吸电子基团强度、引入多位点协同)以及适配大面积工艺,该策略有望在钙钛矿/硅、全钙钛矿及钙钛矿/有机叠层等领域发挥更广泛的作用,推动第三代光伏技术迈向商业化。
文献来源
Wang, S., Wang, Q., Lu, X., Xue, T., Yang, J., He, R., Gao, X., Pan, J., Sun, X., Chen, J., Song, J., Cheng, J., Li, G., Song, Y., & Zhang, Y. (2026). Electron‐Deficient Amines Enable Halide‐Anchoring Hydrogen Bonding for Stable Wide‐Bandgap Perovskites Toward Perovskite/Organic Tandem Solar Cells.Angewandte Chemie International Edition.
https://doi.org/10.1002/anie.9708831
仅用于学术分享,如有侵权,请联系删除。