亲爱的读者们,不星标《纵横科研》公众号,会收不到我们的最新推送点击公众号主页右上角,星标《纵横科研》,不错过每一条科研资讯Breaking Scaling Relations for Selective N-Phenylhydroxylamine Synthesis on Platinum Catalysts via Oxophilic Metal Alloying-Induced Asymmetric Adsorption of −NO2
https://doi.org/10.1021/acscatal.5c07724在硝基芳烃经羟胺中间体加氢为苯胺的级联反应中,克服由线性标度关系导致的羟胺选择性‑活性权衡仍具挑战。本文将铂与亲氧金属合金化,可打破此类线性标度关系,实现高羟胺产率,而无需传统催化剂所需的添加剂。以铂钴/活性炭为主要模型,实验与密度泛函理论计算表明,硝基芳烃还原为羟胺的高性能归因于其不对称吸附,即一个氮‑氧键与铂位点结合,另一个与钴位点结合,这从空间上解耦了初始氮‑氧键断裂与含氮‑氧中间体的后续脱附/加氢,从而打破了硝基苯与N-苯基羟胺间的线性标度关系。在铂位点上,第一个氮‑氧键的断裂以降低的能垒进行,而在钴位点上,N-苯基羟胺的加氢在动力学上不如其脱附有利。这些效应共同作用,在不损失活性的前提下实现了高N-苯基羟胺选择性。铂铁/活性炭和铂镍/活性炭也表现出类似趋势,其性能均优于对应的铂/活性炭催化剂。
芳香硝基化合物的催化氢化是化学工业中的基础转化反应,可用于合成一系列关键中间体。其中,N-苯基羟胺的选择性合成尤为重要,因其是生产高价值精细化学品的关键中间体。然而,高选择性获得该中间体仍是一个重大挑战。催化转化过程涉及硝基苯经亚硝基苯依次还原为N-苯基羟胺和苯胺,其中N-苯基羟胺在热力学上不稳定,易于进一步加氢生成更稳定的苯胺。羟基胺的工业化生产通常需要高活性的氢供体,以在低温下促进氢化反应,从而提高产率。然而,这些氢供体成本高昂,且伴随诸多副产物。因此,开发以氢气为氢供体的替代氢化系统,对于降低制造成本和简化分离过程具有重要意义。尽管如此,该策略仍具挑战性,因为氢气活化通常需要较高温度,而这易于促进N-苯基羟胺过度加氢生成热力学稳定的苯胺。
贵金属基催化剂,特别是铂基体系,极具前景,因其可在温和条件下实现氢气活化,从而减轻高温诱导的过度加氢反应。通常,铂因其d轨道与芳香环π电子的显著轨道重叠,对芳香硝基化合物表现出强吸附,而硝基在铂位点上采取对称的双齿桥接构型。该吸附构型直接活化了整个硝基官能团,这有利于硝基加氢为羟胺,但也阻碍了羟胺中间体的脱附。这种情形不可避免地驱动氮氧键的完全氢解,导致苯胺生成。
传统策略,如配体修饰和载体工程,旨在减弱芳香环的吸附,从而使硝基化合物以相对较弱的相互作用通过硝基吸附在铂催化剂表面。一旦硝基被加氢为羟胺,其更可能从铂催化剂上脱附,而非继续进行连续加氢。然而,仅减弱苯环的吸附并不能解决选择性与催化活性之间的固有平衡。由于含氮氧中间体之间存在线性标度关系,该方法常导致整体催化性能下降。具体而言,硝基加氢生成羟胺涉及多个氮氧键断裂步骤,其中含氮氧中间体表现出线性标度关系。这意味着弱的硝基吸附有利于生成的羟胺脱附,而不是其进一步加氢为苯胺,反之亦然。然而,这种弱吸附无法活化硝基,导致催化活性低下。因此,打破硝基的对称吸附是高效氢化硝基苯制备N-苯基羟胺的关键。
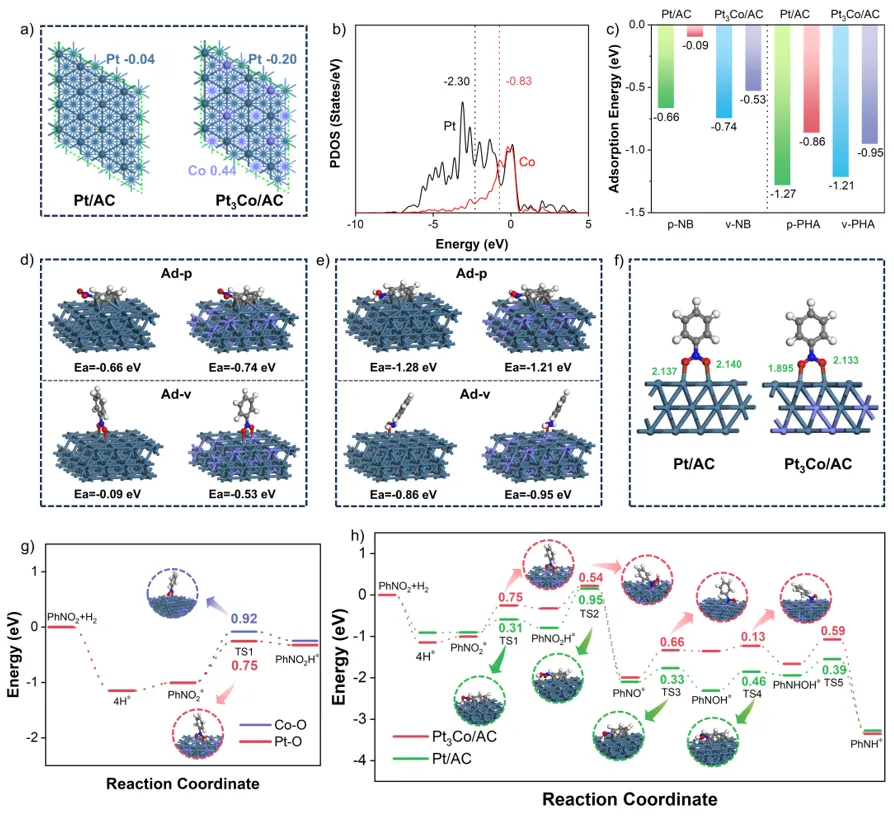
理论上,打破铂连续位点可实现硝基苯中硝基的不对称偶联。该构型允许硝基的两个氧原子吸附在不同位点上,从而在反应过程中使不同含氮氧中间体的吸附位点在空间上解耦。理想情况下,一个位点促进强吸附并启动硝基氮氧键的氢解,而相邻位点对生成的羟胺中氮氧基团表现出弱吸附,从而促进N-苯基羟胺的脱附。因此,将铂与亲氧金属合金化,为规避上述情况提供了潜在解决方案。一方面,将异质金属原子引入原始金属晶格,可能实现对硝基两个氧原子的不对称吸附,打破底物与中间体之间的线性标度关系。另一方面,亲氧金属具有适中的d带中心,可增强对硝基氧的吸附,同时减弱对苯环的吸附,从而避免氢化过程中引入的d‑π相互作用。尽管潜力巨大,但该研究领域的报道相对较少。
本研究证明了将铂与亲氧金属合金化以打破硝基芳烃在铂基催化剂上加氢生成N-苯基羟胺的线性标度关系的可行性,从而实现N-苯基羟胺的高效选择性生产。具体而言,以铂钴/活性炭为模型,研究表明铂钴合金位点促进了硝基的空间不对称吸附,其中一个氮氧键在铂位点形成,另一个在钴位点形成。这种独特的吸附构型促进了硝基第一个氮氧键的断裂,使后续加氢步骤在另一个位点发生,有效打破了N-苯基羟胺与硝基苯之间的线性标度关系。此外,这种情况显著降低了速率控制步骤的能垒,提升了氢化活性。同时,在这些位点上N-苯基羟胺进一步加氢的能垒显著增加,而N-苯基羟胺的脱附能降低,从而实现了对N-苯基羟胺的高选择性。该合金化策略可推广至其他亲氧金属,所得铂‑亲氧金属催化剂可在温和条件下应用于多种硝基化合物。
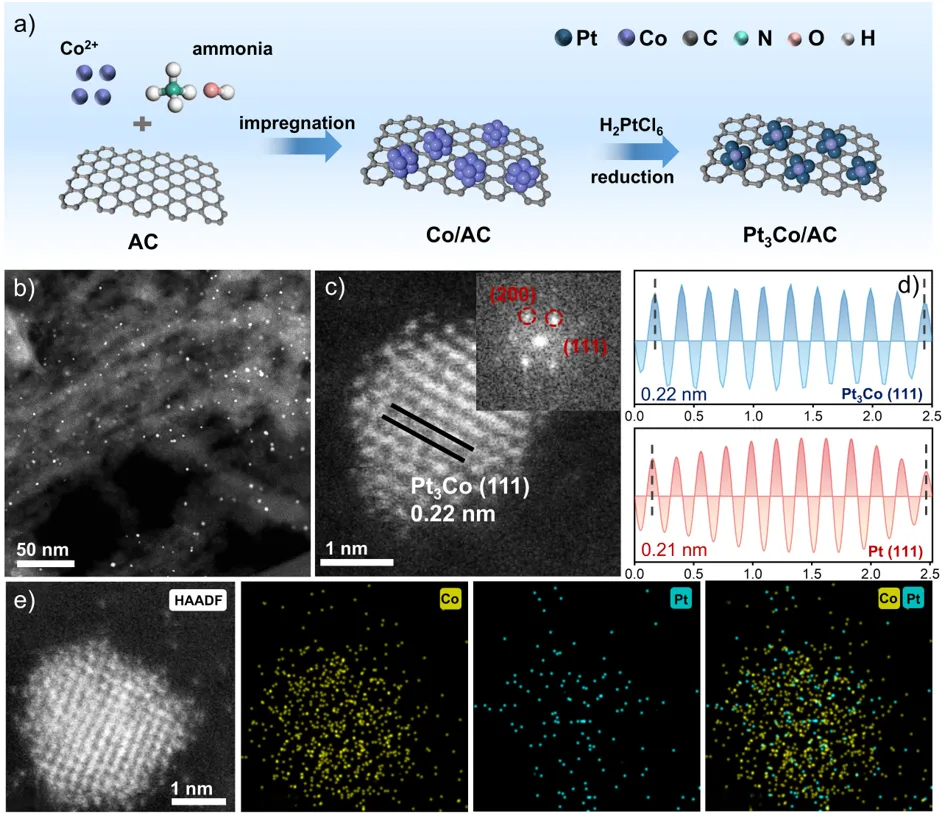
图1. (a) Pt₃Co/AC催化剂的制备示意图。(b) 透射电镜图像。(c) 高分辨透射电镜图像及其快速傅里叶变换衍射图。(d) Pt₃Co/AC与Pt/AC催化剂的逆快速傅里叶变换图像。(e) 原子分辨高角环形暗场扫描透射电镜图像。(f) Pt₃Co/AC催化剂的钴、铂及混合区域元素能量色散X射线光谱分布图。
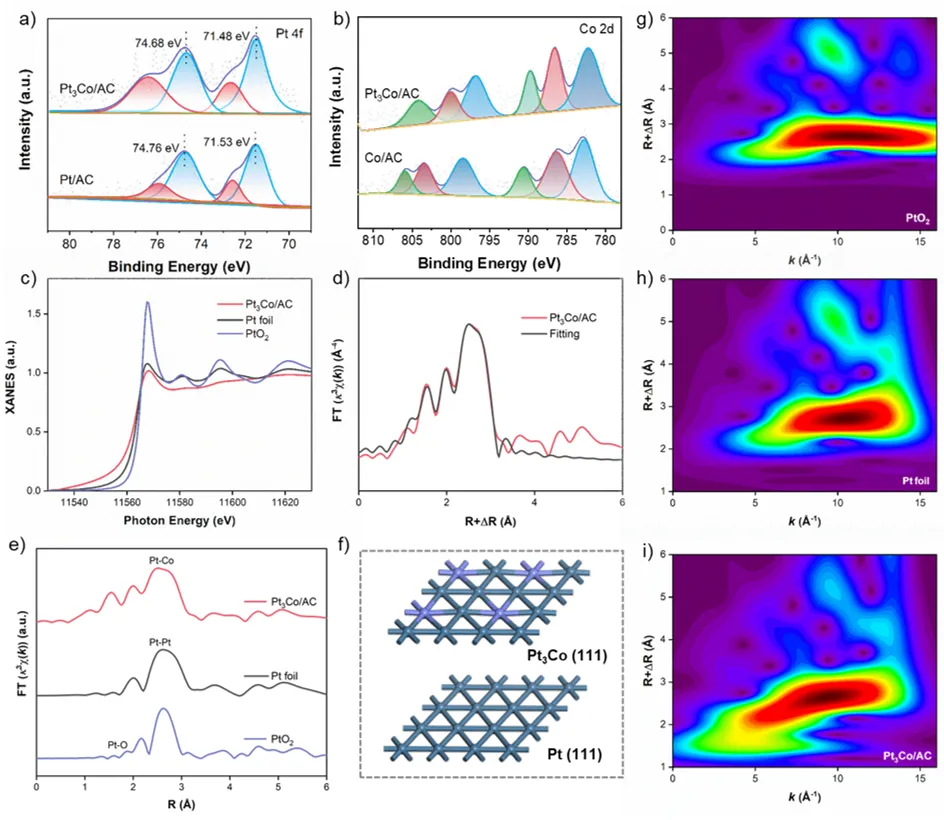
图2. (a) 铂4f轨道与(b) 钴2p轨道X射线光电子能谱。(c) X射线吸收近边结构谱。(d) Pt₃Co/AC的扩展X射线吸收精细结构谱拟合曲线。(e) Pt₃Co/AC的铂L₃边傅里叶变换扩展X射线吸收精细结构谱。(f) Pt₃Co(111)与Pt(111)晶面的结构示意图。(g–i) Pt₃Co/AC、PtO₂与铂箔的k³加权扩展X射线吸收精细结构谱的小波变换图。
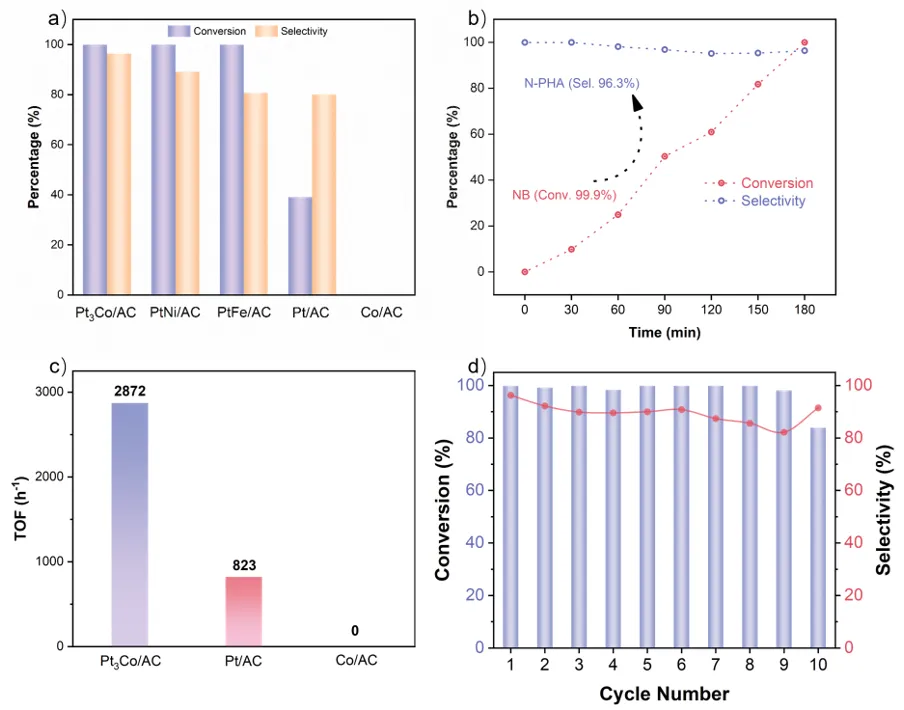
图3. (a) Pt₃Co/AC、Pt/AC与Co/AC的催化性能。(b) Pt₃Co/AC上硝基苯氢化的动力学曲线。(c) Pt₃Co/AC、Pt/AC与Co/AC的转换频率。(d) Pt₃Co/AC催化剂的氢化稳定性测试
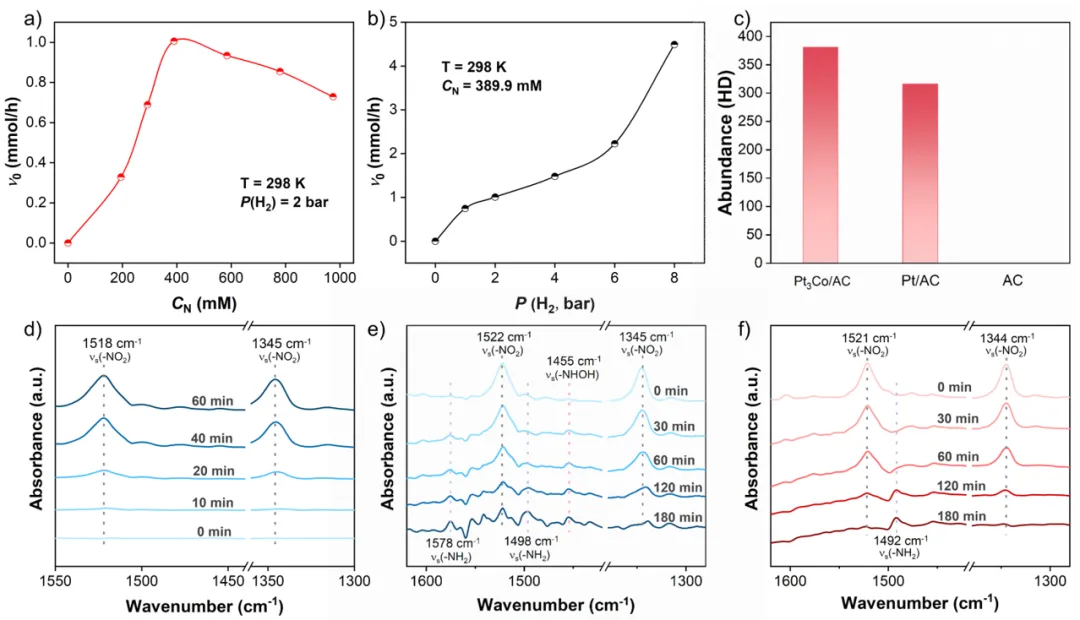
图4. (a) 硝基苯浓度与(b) 氢气压力对初始氢化反应速率的影响。(c) 氢气‑氘气交换实验结果。硝基苯在Pt₃Co/AC上吸附的(d) 原位漫反射红外傅里叶变换光谱,及(e, f) Pt₃Co/AC与Pt/AC催化剂上还原过程的原位漫反射红外傅里叶变换光谱。
图5. (a) Pt/AC与Pt₃Co/AC的Bader电荷分析。(b) Pt₃Co/AC的总态密度图。(c) Pt₃Co/AC与Pt/AC上硝基苯和N-苯基羟胺的吸附能垒。(d) 硝基苯在Pt/AC与Pt₃Co/AC上的吸附模型。(e) N-苯基羟胺在Pt/AC与Pt₃Co/AC上的吸附模型。(f) 硝基苯在Pt/AC与Pt₃Co/AC上的吸附键长。(g) Pt₃Co/AC上第一步氮氧键断裂中铂‑氧与钴‑氧键的能垒对比。(h) Pt/AC与Pt₃Co/AC上硝基苯氢化为苯胺的能量路径。
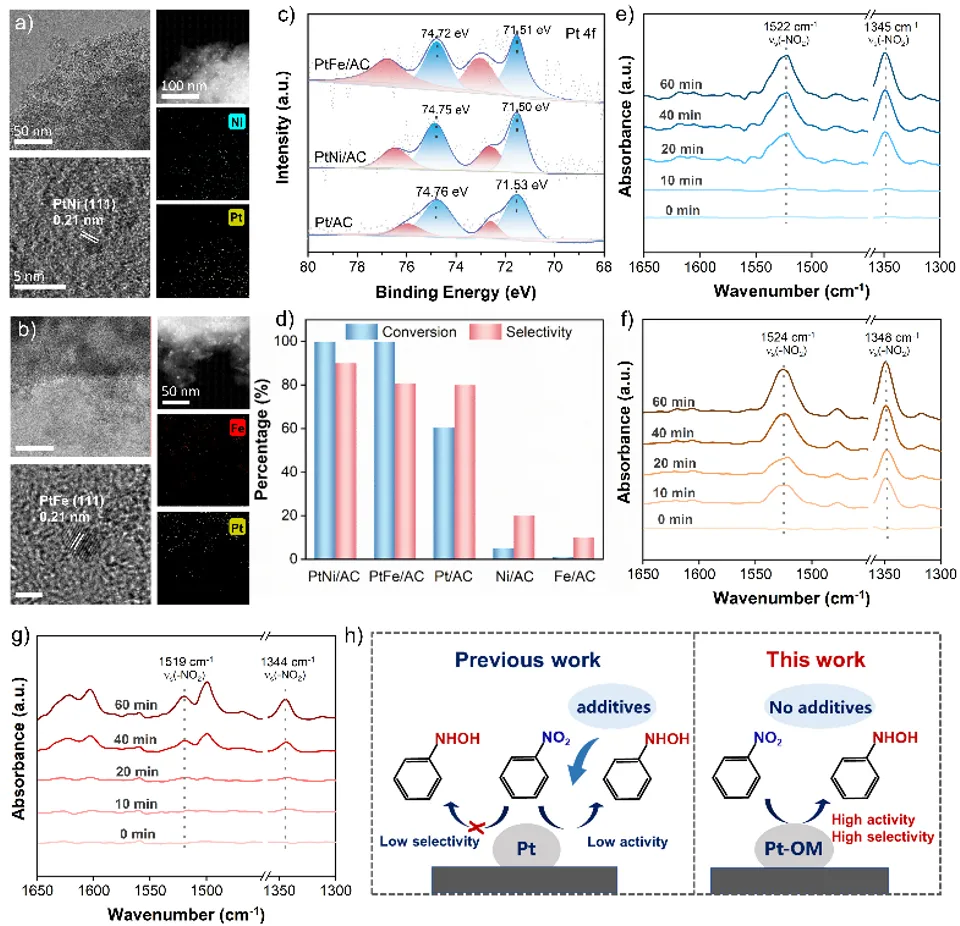
图6. (a) PtNi/AC与(b) PtFe/AC催化剂的透射电镜图像、高分辨透射电镜图像、高角环形暗场扫描透射电镜图像,及钴、铂元素能量色散X射线光谱分布图;(c) PtNi/AC、PtFe/AC与Pt/AC催化剂的铂4f轨道X射线光电子能谱;(d) PtNi/AC、PtFe/AC、Ni/AC、Fe/AC与Pt/AC的催化性能对比。(e–g) PtNi/AC、PtFe/AC与Pt/AC催化剂上硝基苯吸附的原位漫反射红外傅里叶变换光谱。(h) 本工作与其他工作的性能对比。
总之,将铂与亲氧金属合金化,打破了硝基芳烃催化氢化合成N-苯基羟胺中因吸附标度关系所导致的活性‑选择性限制。与铂/活性炭或商用铂/碳催化剂相比,铂钴/活性炭表现出优异的N-苯基羟胺选择性。实验与理论分析表明,与亲氧金属合金化诱导了硝基在铂‑亲氧金属位点的不对称吸附。这种独特的吸附构型使得硝基苯加氢过程中不同氮氧键的断裂发生在不同的位点,有效打破了氢化过程固有的线性标度关系。该策略不仅降低了氢化反应速率控制步骤的能垒,也抑制了N-苯基羟胺的进一步氢化,从而克服了催化氢化中的活性‑选择性权衡。本工作不仅提供了一种高效催化剂,而且深化了对硝基苯氢化制备N-苯基羟胺机理的理解,有望为其他类型级联氢化催化剂的理性设计带来启发。