LIB-Raman丨郑州大学陈卫华教授Nat.Commun.:原位表征揭示双域溶剂锁定型电解液中阴离子优先分解与界面钝化机制
提升钠离子电池的截止电压上限有助于实现电极容量最大化,并缩小其与商用锂离子电池之间的能量密度差距。传统电解液在高度脱钠正极表面不稳定,易受亲电攻击并持续分解,形成富含低聚物的界面相以及容量快速衰减。本文设计了一种溶剂锁定型碳酸酯基电解液,形成电化学稳定的溶剂增强型溶剂化结构,并在正极上形成由阴离子富集界面屏蔽层衍生的富含硼化物/氟化物的界面相,这种电解液与商业氧化物及聚阴离子正极材料具有良好的兼容性。Na||Na₂.₂₆Fe₁.₈₇(SO₄)₃电池在4.5 V,1000 mA g⁻¹电流密度下循环16500次后容量仍保持88.2%(扣式电池);在100 mA g⁻¹下循环500次后容量保持93.9%(软包电池)。
在钠离子电池的高度脱钠正极表面,电子结构变化导致正极呈强亲电性,容易攻击电解液中的自由溶剂分子和钠离子溶剂化络合物。由溶剂主导的分解会产生有机自由基,并通过亲电加成-缩聚反应形成富含低聚物的多孔界面相,导致电解液持续消耗、容量快速衰减。常规碳酸酯电解液在高电压下会引发副反应和安全问题。目前的方法多为单方面调控,例如通过高浓度或局部高浓度电解液调控体相溶剂化结构,或使用含氟添加剂预先形成界面膜。这些方法在实际应用中造成离子电导率低、粘度增加,或与负极不兼容。本研究设计了一种双域溶剂锁定型电解液(DDSLE),在1 M NaPF₆ EC/EMC中加入二氟草酸硼酸根阴离子(DFOB-)。DFOB-与暴露的Na⁺正极表面强结合,利用其空间位阻和吸电子效应,在体相电解液(域I)和正极界面(域II)“锁定”溶剂分子。
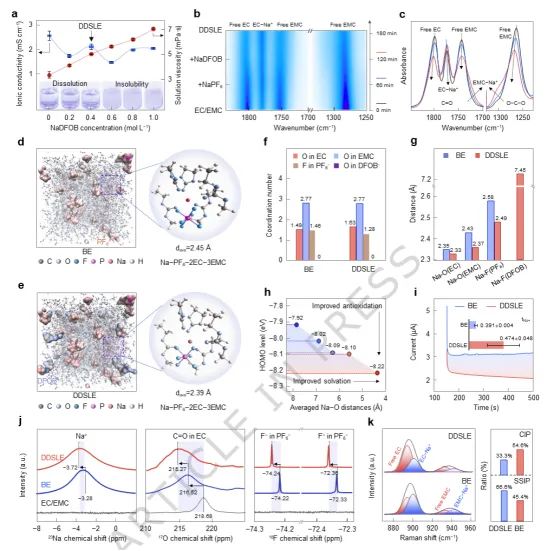
图1. DDSLE电解质的化学和溶剂化结构。(a) 不同NaDFOB浓度下电解液的离子电导率、溶液粘度。(b,c) 从BE电解液到DDSLE电解液,EC/EMC中盐溶解过程的原位FTIR光谱。(d) BE和(e) DDSLE电解液的分子动力学模拟,PF₆⁻用黄色等值面表示,DFOB⁻用蓝色等值面表示;Na⁺离子以及EC/EMC分子以棍棒模型显示。(f) MD模拟的平均配位数、(g) Na–O/F键长。(h) Na⁺溶剂化结构的HOMO能量随配位距离的变化。(i) BE和DDSLE电解液在Na||Na对称电池中的计时电流曲线,插图为Na⁺迁移数。(j) 纯溶剂、BE电解液和DDSLE电解液的²³Na、¹⁷O和¹⁹F核磁共振谱。(k) BE和DDSLE电解液的拉曼光谱,以及相应的溶剂化物种定量结果。选择1 M NaPF₆与0.4 M NaDFOB溶于EC/EMC溶剂中作为DDSLE电解质的最优配比,平衡电导率与稳定性(图1a)。采用原位傅里叶变换红外光谱探究制备DDSLE过程中的溶剂化结构重组机制(图1b-c)。在DDSLE电解质中,自由组分蓝移:EC的C=O峰从1800 cm⁻¹移至1805 cm⁻¹,EMC的C–O–C峰从1270 cm⁻¹移至1274 cm⁻¹,表明体系的电子环境发生改变且分子间相互作用增强。这些谱峰位移源于溶剂氧原子孤对电子向Na⁺空轨道的电子供体作用,削弱了氧原子的电子云密度,增强了化学键力常数,导致振动频率升高。
添加NaDFOB后,Na⁺的溶剂化壳层组成保持相似(图1d–e),PF₆⁻的配位数从1.46降至1.28,EC的配位数从1.49升至1.63,而EMC的配位数保持2.77(图1f)。DFOB⁻阴离子表现出强烈的非溶剂化效应,压缩了初始溶剂化壳层。相应地,Na⁺与相邻分子间的关键键长均缩短:Na–O(EC)从2.35 Å缩短至2.33 Å,Na–O(EMC)从2.43 Å缩短至2.36 Å,Na–F(PF₆⁻)从2.58 Å缩短至2.48 Å(图1g)。密度泛函理论计算显示,溶剂化结构的HOMO能级从–7.92 eV降至–8.22 eV,抗氧化稳定性提高(图1h)。计时电流法测得Na⁺迁移数从BE的0.391提升至DDSLE的0.474(图1i)。核磁共振谱证实DDSLE电解质中溶剂强化的溶剂化结构,Na⁺周围电子密度增加、溶剂配位作用增强(图1j)。拉曼光谱分析显示,BE电解质中以接触离子对为主(CIP,33.3%),而DDSLE电解质中溶剂分离离子对(SSIP,45.4%)比例显著提高(图1k)。这源于DFOB⁻阴离子强烈的吸电子效应与空间位阻特性,增强了局部电场,驱动电子密度重分布,稳定了溶剂强化的溶剂化结构,从而提升电解质的稳定性和传输性能。
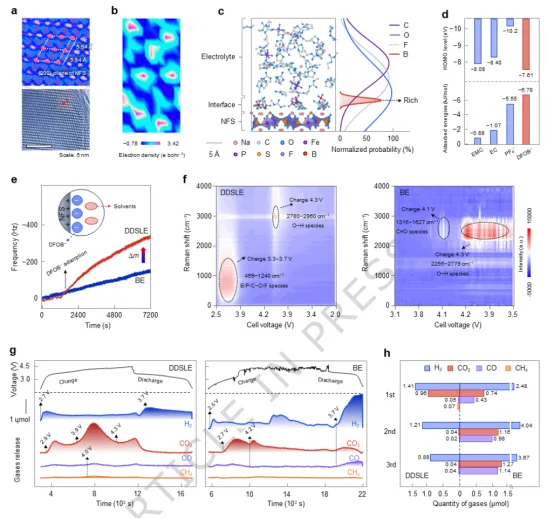
图2. DDSLE电解质在正极界面的结构演变与反应过程。(a) NFS正极的高分辨透射电子显微镜图像。(b) NFS(200)晶面的电子密度。(c) NFS/DDSLE界面模型。(d) 电解质组分在NFS表面的吸附能及最高占据分子轨道能级。(e)在50 mV·s⁻¹扫描速率和25℃下测得Na||NFS电池频率随时间变化曲线。(f) 采用DDSLE与BE电解质的Na||NFS电池在50 μA·cm⁻²和25℃下的原位拉曼光谱。(g) 使用DDSLE和BE电解质的Na||NFS电池在50 μA·cm⁻²和25℃下首次充电至4.5 V过程的原位差分电化学质谱。(h) 气体释放量。
DDSLE电解液在正极界面处的结构演化如图2所示。橄榄石型Na₂.₂₆Fe₁.₈₇(SO₄)₃(NFS)正极在2.0–4.5 V电压范围内具有较高的平均放电电压和可逆比容量。HRTEM分析证实,具有规则单斜晶系原子排列的(200)晶面为主要暴露面(图2a)。电子密度分布显示,配位氧位点存在局域化高密度区域,而由于Na含量高,表面整体呈电负性(图2b)。分子动力学模拟表明,与PF₆⁻、EC和EMC相比,DFOB⁻具有更强的吸附能,形成致密的富阴离子的界面屏蔽层,隔离正极与电解质(图2c–d)。沿c轴的组分分布图证实,DFOB⁻在正极/电解质界面附近富集。EQCM测试显示,NFS颗粒与DDSLE电解质接触1309 s后,其负质量谐振频率快速下降(图2e),证明DFOB⁻吸附过程加速以及形成富阴离子界面屏蔽层。DFOB⁻的最高占据分子轨道能级最高,氧化稳定性低于其他组分(图2d),能够在正极表面优先氧化。原位拉曼光谱显示,在初始充电过程中,在469–1240 cm⁻¹范围内出现新峰,对应DFOB⁻氧化分解形成B/P/C–O/F物种(图2f)。充电至4.2 V时,2780–2960 cm⁻¹范围内的峰变化表明富H–O物种从溶剂分解。相比之下,采用BE电解质的电池在超过3.9 V后,在1316–1627 cm⁻¹出现明显的峰,并在4.2 V以上在2255–2779 cm⁻¹出现强峰,证实溶剂在更低的电压下发生严重分解。原位差分电化学质谱印证该分析,对于使用DDSLE的电池,CO2释放始于2.9 V(充电阶段),在3.9 V增强,短暂降低后在4.3 V以上再次上升,而CO/CH4的释放量极少(图2g),表明在溶剂分解前,DFOB⁻分解是CO2的主要来源。使用BE的电池由于界面结构未受屏蔽,CO2释放起始更早,且在放电阶段出现CO2/CO的重复峰。多循环DEMS定量分析显示(图2h),DDSLE仅在首次循环中产生气体。而基于BE的电池不仅在首次循环产生大量气体,且在后续循环中气体生成量持续增加。以上结果证明,源自富阴离子屏蔽层的钝化层能抑制高压下电解液的持续分解。DFOB⁻优先氧化将电解质的阳极稳定性从3.4 V vs.Na⁺/Na(BE电解质)提升至5.1 V vs.Na⁺/Na(DDSLE电解质)。
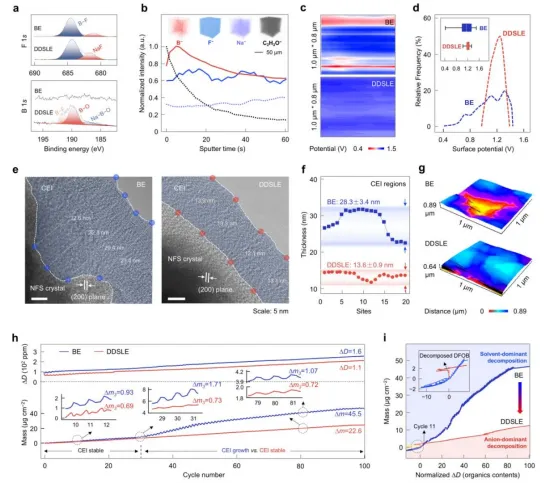
DDSLE电解液在NFS正极上形成的正极电解质界面相的组成、结构与力学性能如图3所示。XPS分析表明(图3a),DDSLE生成的CEI富含硼化物和氟化物;而BE生成的CEI几乎不含硼,且富含有机物种。TOF-SIMS深度剖析表明(图3b),DDSLE的CEI呈分层结构:B在表层富集,F⁻和Na⁺分布均匀,C₂H₅O⁻仅在最表层。DDSLE的CEI表面电势均匀,电荷耗散能力强;BE的CEI电势分布不均,存在局部电荷积累,如图3c-d所示。NFS正极循环后的cryo-TEM图像显示出对应于(200)晶面的清晰晶格条纹,表明其整体结构良好(图3e-f)。BE电解质形成厚且不均匀的CEI,而DDSLE形成显著更薄且均匀的CEI。AFM表征证实(图3g),DDSLE衍生的界面膜更光滑和更均匀,有利于离子的快速跨界面传输。EQCM测试阐明了100次循环期间DDSLE衍生CEI的机械稳定性(图3h)。DDSLE的CEI在30次循环后趋于稳定;BE的CEI质量持续快速增长,表明CEI破裂和电解液持续分解。DDSLE的质量变化与耗散比值低且平稳,对应刚性、无机组分为主的CEI;BE的比值高且上升,对应软而多孔的有机CEI(图3i)。
图3.CEI的成分、结构与机械性能。(a) NFS正极循环后的B 1s与F 1s XPS谱图。(b) DDSLE衍生的CEI中B⁻、F⁻、Na⁻和C₂H₃O⁻物种的TOF-SIMS三维重构及其相对强度随溅射深度变化。(c,d) 使用DDSLE和BE电解质的循环后NFS正极的静电势分布图。(e) 使用BE与DDSLE电解质所形成的CEI的冷冻透射电子显微镜图像。(f) CEI厚度分布。(g) 使用DDSLE和BE电解质的循环后NFS正极表面粗糙度映射图。(h) 通过原位EQCM记录在50 mV s⁻¹和25°C条件下进行CV测试时,Na||NFS电池在多个循环过程中质量变化(Δm)与能量耗散(ΔD)随时间的变化曲线。
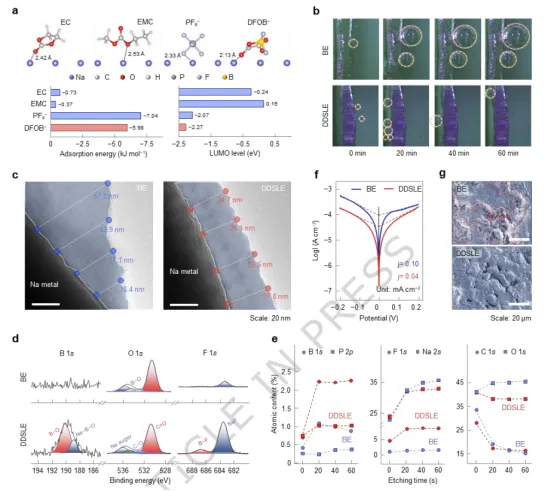
图4.界面化学与SEI结构。(a) 电解质组分在Na金属表面的吸附结构、吸附能及LUMO能级。(b) 使用DDSLE与BE电解质的Na||Na对称电池在50 μA cm⁻²和25°C条件下,Na金属表面的原位光学显微镜图像。(c) 在BE和DDSLE电解质中形成的SEI的透射电子显微镜图像。(d) 循环后Na负极的B 1s与F 1s XPS谱图。(e) 元素组成随XPS溅射深度的演变。(f) 采用DDSLE和BE电解质的Na||Na对称电池的Tafel曲线。(g) Na||NFS电池循环500次后Na金属表面的SEM图像。
DDSLE电解液在Na负极表面形成的固体电解质界面相的化学组成、结构与稳定性如图4所示。DFOB⁻在Na金属表面表现出较强的吸附能,而溶剂的吸附能为−0.37~−0.73 kJ mol⁻¹。结合其较低的LUMO能级,DFOB⁻易于从Na负极接受电子,从而优先还原分解(图4a)。对称Na电池的光学显微镜图像显示,在Na沉积/剥离的初始20分钟内出现致密微气泡,对应DFOB⁻还原(图4b)。这些气泡随后消散且未持续增长,证实界面能快速稳定。相比之下,BE电解质在裸露的Na金属表面持续产生气泡。初始循环五次后,DDSLE电解质中的SEI更均匀(图4c)。XPS分析表明,DDSLE衍生的SEI富含硼物种和氟化物,而基于BE的SEI则未检测到硼,且NaF含量极低(图4d)。深度剖析显示,在DDSLE中,硼含量在最外层表面相对较低(约0.68%),但在蚀刻后急剧上升并稳定在约2.3%(图4e)。DDSLE体系的腐蚀电流密度(0.04 mA cm⁻²)低于BE体系(0.10 mA cm⁻²),表明其界面钝化效能显著增强(图4f)。循环后的SEM图像证实,DDSLE电解质可实现可逆的Na沉积:经过500次循环后Na箔仍保持光滑表面;相比之下,BE电解质中Na表面出现苔藓状沉积(图4g)。
原位质谱测试实验细节
在充满Ar的手套箱中,使用北京中研环科科技有限公司(https://www.bjscistar.com)设计的原位模具组装电池,正极的活性物质负载量为1.0–1.5 mg cm⁻²,Na金属为对电极,采用直径为16 mm的玻璃纤维隔膜,电解液200 μL,在50 μA cm⁻²下进行充放电测试。载气为预脱水的高纯He(99.999%);气体管路先以高流速吹扫以除去残留空气,然后通过质量流量控制器将流速稳定在1 mL/min。电池产生的气体先经过一个低温冷凝器,捕集电解液中的挥发性组分,随后由HPR-20型微分电化学质谱系统进行检测。
原位EQCM测试实验细节
将NFS粉末分散于PVDF/NMP溶液(质量分数3%,5 mL)中,形成悬浊液。超声处理30分钟后,取20 μL上清液滴涂到硅片上,并在120°C下真空干燥4小时。在充满Ar的手套箱中使用专用模具组装原位电池,以Na金属为负极,并加入200 μL电解液。采用QSense Explorer型电化学石英晶体微天平进行EQCM测试,使用镀金石英晶体传感器。镀金传感器作为工作电极;充放电循环采用恒电位阶跃模式,扫描速率50 mV s⁻¹,电压范围2.0–4.5 V vs.Na⁺/Na,循环次数100次。
原位Raman测试实验细节
将活性物质载量为1.0–1.5 mg cm⁻²的正极与钠金属对电极、玻璃纤维隔膜一同组装于由北京中研环科科技有限公司(https://www.bjscistar.com)设计的原位电池中,电池内加入200 μL电解液。使用HORIBA Scientific Lab-RAM HR Evolution型拉曼光谱系统采集光谱,激发波长532 nm,扫描间隔10分钟。充放电过程由Lvium电化学工作站控制,电流密度为50 μA cm⁻²。
所有原位表征均在恒温环境(25±1°C)下进行。
本研究提供了一种双域溶剂锁定型电解液(DDSLE),以解决工业化NaPF₆/碳酸酯基电解液在高电压(>4.3 V)下长期不稳定的挑战。通过引入对称的二氟草酸硼酸根阴离子(DFOB⁻),在体相溶液(域I)中构建了溶剂增强的Na⁺溶剂化结构,并在正极界面(域II)处形成阴离子富集屏蔽层,有效限制溶剂分子的自由度和反应活性。这种溶剂锁定结构将电解液的分解途径从溶剂主导转变为阴离子主导,从而促进形成稳定的界面相。在正极侧,形成了一种内层富含硼化物/氟化物、外层富含聚酯/醇盐的集成式正极电解质界面相,有效抑制电子隧穿,使正极接触电势降低16.7%,漏电流降低34.6%–79.2%。实验与理论分析确定了电子隧穿电阻是高电压界面稳定性的主要决定因素。带隙超过6.2 eV的硼化物/氟化物能够有效抑制4.0 V以上的电子隧穿。同时,在Na负极上形成了薄而均匀的SEI,实现可逆Na沉积。这些协同效应提高了电解液在高电压下的稳定性,表现为阳极稳定性极限提升、库仑效率提高、过充现象及气体释放减少。该DDSLE电解液与钠负极以及多种正极材料均展现出良好的兼容性。Na||Na₂.₂₆Fe₁.₈₇(SO₄)₃电池在4.5 V截止电压下表现出优异的长期循环稳定性:扣式电池在16500次循环后容量保持率为88.2%,软包电池在循环500次后容量保持率为93.9%。
Zhang, J., Tang, G., Ma, S. et al. Dual-domain solvent-locked electrolyte enabled durable 4.5 V-class sodium batteries. Nat Commun (2026).
DOI:10.1038/s41467-026-72849-z
https://doi.org/10.1038/s41467-026-72849-z
我们拥有一支由教授、博士领衔的技术研发团队,在同步辐射技术以及应用,高温高压实验技术,高温真空设备研制方面具有多年的实验和系统集成经验,拥有多项专利及知识产权,可以根据老师实验要求按需定制,力求为您提供更专业的服务!现已和国内各大科研院所及高校达成广泛的合作,中科院高能所,金属所,物理所,上海光源,合肥光源等,有中科大,北大,清华,吉大,复旦,上交,南大,浙大,武大,中山大学等等。更多产品欢迎登陆网站了解。http://www.bjscistar.com
更多原位信息
请长按下方图片
识别二维码 关注北京中研环科
电话:010-83601911
手机:19392795346(微信同)
网址:www.bjscistar.com
邮箱:hp@bjscistar.com