定制O-O自由基偶联路径,用于高密度Fe−N−C催化剂,实现工业级水的稳定氧化
第一作者:胡志昂
通讯作者:卢思宇教授、常江伟研究员、唐志勇院士
单位:郑州大学、中国科学院
链接:https://doi.org/10.1002/anie.9504269
阴离子交换膜水电解(AEMWE)结合了碱性水电解的低成本、高能量效率与质子交换膜水电解的高电流密度、高氢气纯度优势,是极具前景的大规模制氢技术。然而,AEMWE 技术的实际应用面临重大挑战,特别是在工业级高电流密度(≥5 A cm-2)下,缓慢的析氧反应(OER)动力学和不足的长期运行稳定性严重限制了其性能和大规模部署。目前主流的先进系统通常只能在低于5 A cm-2的电流密度下运行,基于传统材料的AEMWE器件往往只能维持数小时的使用寿命。单原子催化剂(SACs)结合了均相和多相催化的结构与功能优势,在水电解电催化领域展现出巨大潜力。但在实际器件制备中,传统M-N-C单原子催化剂普遍存在金属负载量低(通常< 5 wt.%)的瓶颈,需要厚催化层才能获得足够的催化活性,这会显著加剧传质阻力,导致过电位升高和稳定性下降。此外,稀疏分布的金属原子形成空间孤立的活性位点,使得OER只能遵循固定的吸附演化机制(AEM),受限于强线性标度关系,无法实现更高效的反应路径。
近期提出的氧化物路径机制(OPM)通过相邻*O中间体直接偶联形成O─O键,能够突破传统AEM的线性标度限制,同时避免晶格氧机制(LOM)导致的结构降解和金属流失问题。但如何精准调控单原子催化剂的活性位点密度,实现OER路径从AEM到OPM的可控转变,仍是领域亟待解决的核心科学问题。
近期,郑州大学和中国科学院的研究团队在Angewandte Chemie International Edition发表了题为“Customized O─O Radical Coupling Route on High-Density Fe─N─C Catalysts for Stable Industrial-Scale Water Oxidation”的研究论文。该研究通过理论计算揭示了Fe位点密度对Fe─N─C单原子催化剂上OER关键中间体吸附能的连续调控效应,实验上通过高氮辅助硫化物介导原子捕获策略,制备出Fe负载量高达14.7 wt.%的高密度单原子催化剂(HDFe-N-C),实现了OER机制从AEM到OPM的可控转变。该催化剂在三电极体系中,500 mA cm-2电流密度下过电位仅288 mV,可稳定运行2000 h;组装成阴离子交换膜电解槽后,在5 A cm-2工业级电流密度下电压仅2.15 V,稳定运行500 h,且经过35000次快速启停循环后性能无明显衰减。这项工作为设计高效稳定的非贵金属OER催化剂提供了全新思路,也为单原子催化剂的反应路径工程奠定了理论和实验基础。
要点一:理论预测Fe位点密度对OER机制的调控作用
通过密度泛函理论(DFT)计算,系统研究了不同Fe负载量下Fe─N─C催化剂的电子结构和OER反应路径,结果表明随着Fe位点密度增加,Fe的d带中心从低负载样品的-1.09 eV上移至高负载样品的-1.30 eV,显著增强了表面OH吸附能力。低Fe密度(LDFe-N-C)的孤立活性位点只能遵循AEM路径,受OOH与OH吸附能的线性标度关系限制,反应决速步能垒较高;而高Fe密度(HDFe-N-C)的相邻活性位点能够协同作用,通过O─*O直接偶联形成O─O键,触发热力学更有利的OPM路径,有效降低了反应决速步的能垒。
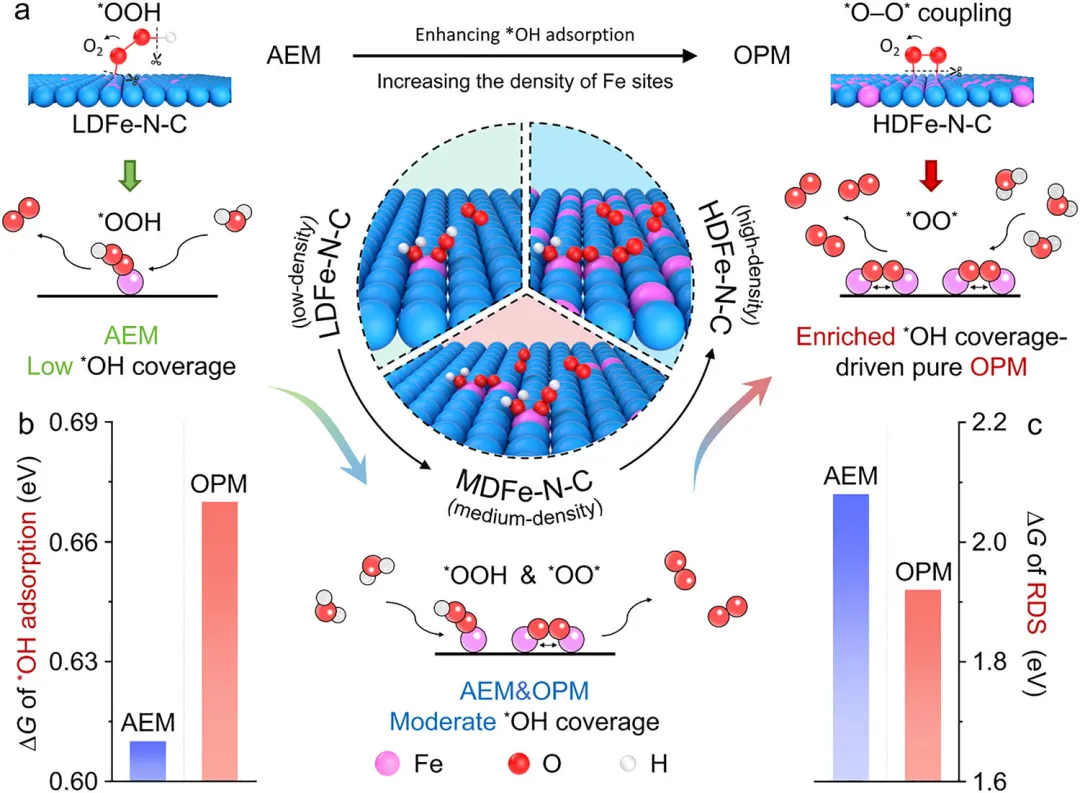
图1 不同Fe位点密度下析氧反应(OER)的反应机制。(a)金属密度调控OER反应路径的示意图。高密度Fe-N-C(HDFe-N-C)表面有利于OH富集,从而触发氧化物路径机制(OPM)。(b) HDFe-N-C与低密度Fe-N-C(LDFe-N-C)上OH吸附的相对能量。(c) HDFe-N-C与LDFe-N-C分别遵循吸附演化机制(AEM)和OPM路径的吉布斯自由能变(ΔG)图对比。基于OPM的HDFe-N-C表现出更低的反应能垒。
要点二:HDFe─N─C单原子催化剂的原子级结构表征
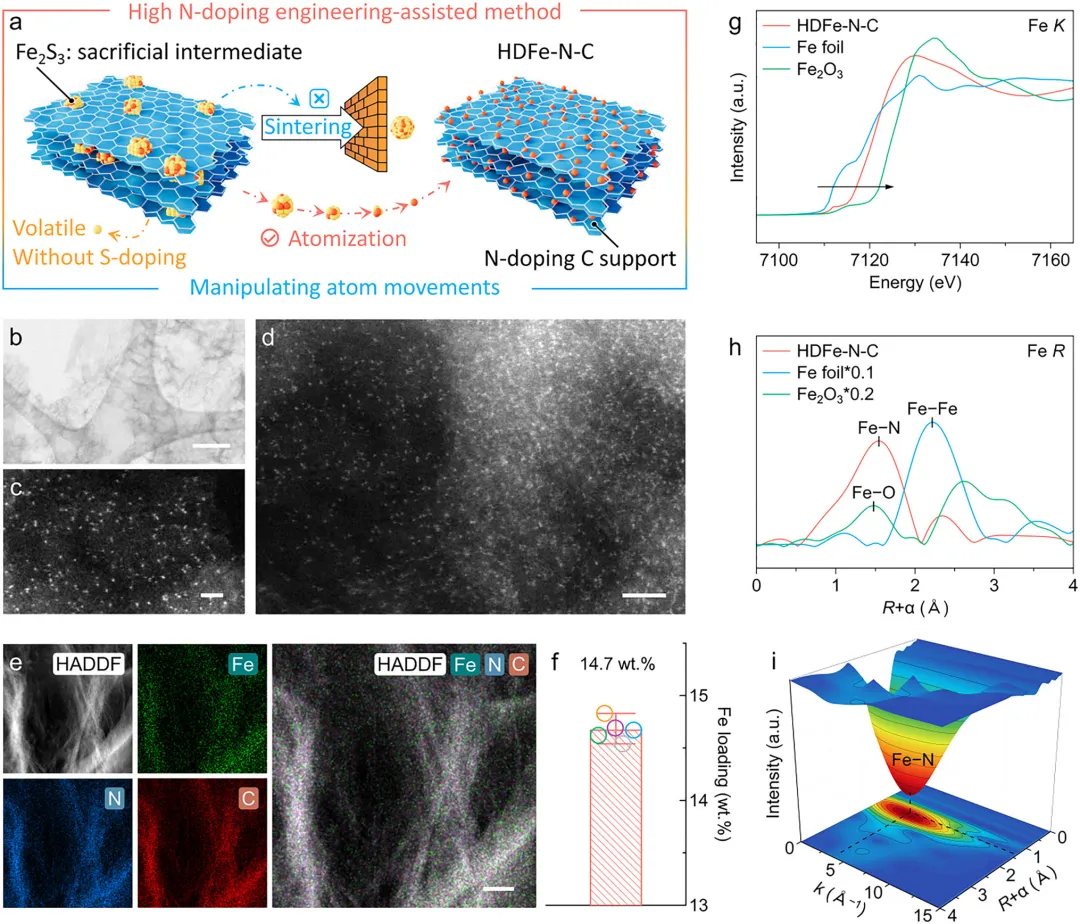
采用高氮辅助硫化物介导原子捕获策略,成功合成了Fe负载量达14.7 wt.%的HDFe-N-C催化剂,通过多尺度表征技术对其原子结构进行了系统分析,球差校正高角环形暗场扫描透射电子显微镜(AC-HAADF-STEM)证实Fe以原子级均匀分散在碳载体上,无团簇或纳米颗粒生成,X射线衍射(XRD)仅观察到石墨碳的特征峰,无金属铁或铁氧化物的衍射信号,X射线吸收精细结构(EXAFS)光谱显示Fe主要以Fe-N4配位结构存在,配位数约为4,X射线光电子能谱(XPS)分析进一步表明Fe以正价态存在,主要与吡啶氮配位。
图2 HDFe-N-C的原子结构与元素分布。(a) 高氮掺杂工程辅助的HDFe-N-C合成策略。(b-d) 球差校正高角环形暗场扫描透射电子显微镜(AC-HAADF STEM)图像,显示HDFe-N-C中Fe原子高度分散且呈孤立状态。(e) HDFe-N-C的环形暗场(ADF)图像及Fe、N、C的局部能量色散X射线光谱(EDX)原子分布图与叠加图。(f) 电感耦合等离子体发射光谱(ICP-OES)测定的HDFe-N-C中Fe的平均掺杂含量。(g) HDFe-N-C及参比材料的归一化Fe k-edge X射线吸收近边结构(XANES)光谱。(h) HDFe-N-C、商业Fe2O3及Fe箔的归一化Fe k-edge傅里叶变换扩展X射线吸收精细结构(FT EXAFS)光谱。(i) HDFe-N-C的k3加权小波变换扩展X射线吸收精细结构(WT-EXAFS)等高线图。比例尺:(b) 200 nm,(c) 1 nm,(d) 2 nm,(e) 100 nm。
要点三:实验验证Fe密度依赖的OER反应路径转变
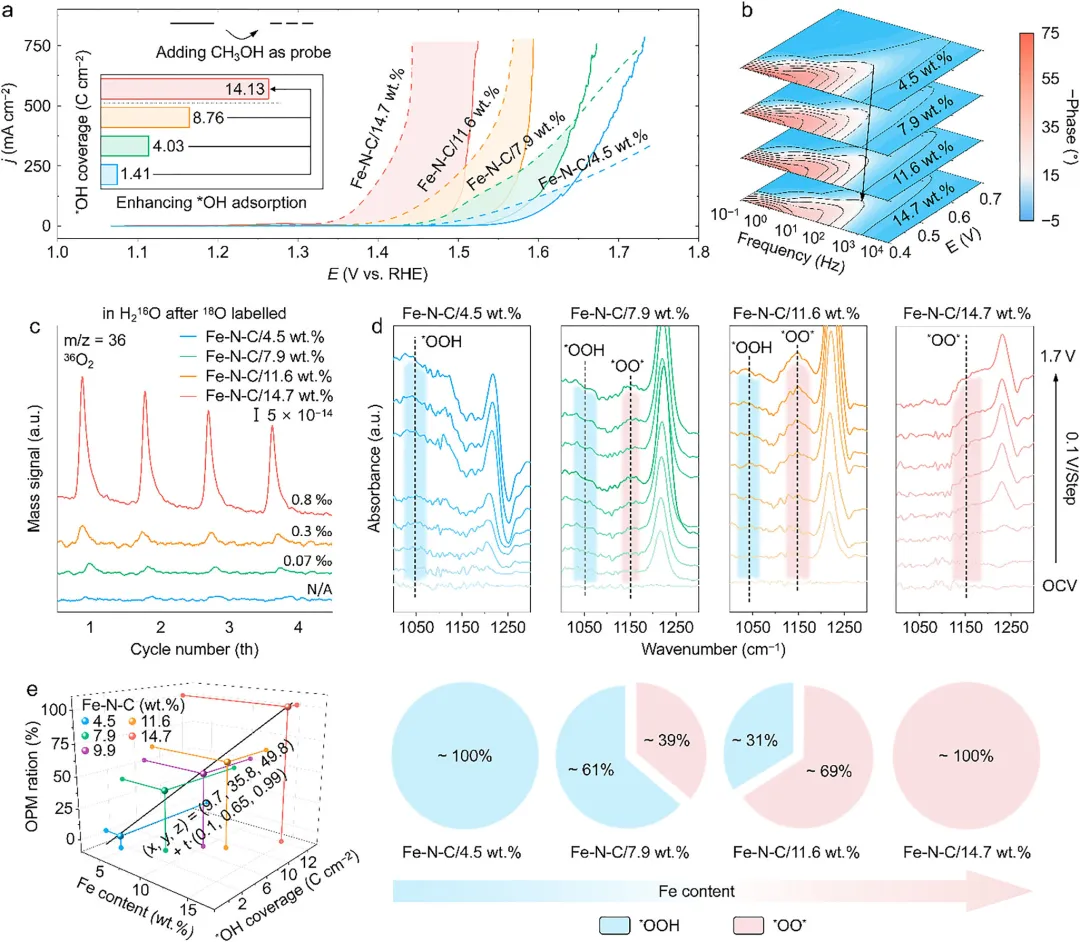
通过一系列原位和非原位表征技术系统验证了Fe位点密度对OER反应路径的调控作用,甲醇氧化探针实验表明随着Fe负载量增加,催化剂表面OH吸附强度和覆盖度显著提高,原位同位素标记微分电化学质谱(DEMS)检测到低Fe负载样品中几乎无36O2(OPM特征信号)生成,而高Fe负载样品中36O2占比显著提升,证明OPM成为主导反应路径,原位衰减全反射表面增强红外吸收光谱(ATR-SEIRAS)显示低Fe负载样品仅出现AEM特征的OOH峰(~1025 cm-1),高Fe负载样品则以OPM特征的*O─O桥接氧峰(~1150 cm-1)为主,定量分析进一步表明Fe负载量、表面OH覆盖度与OPM贡献比例呈良好的线性关系,明确了“密度调控吸附→吸附决定路径”的核心机制。
图3 不同 Fe 负载量 Fe-N-C 催化剂的 OER 机制探究。(a) OER 与甲醇氧化反应(MOR)曲线。插图为极化曲线的电流差值。(b) 原位电化学阻抗谱。(c) 原位微分电化学质谱(DEMS)测得的36O2信号详细对比。(d) 原位衰减全反射表面增强红外吸收光谱(ATR-SEIRAS)(上图)及位于~1025 cm-1和~1150 cm-1处特征峰的强度比变化。(e) 梯度Fe负载量Fe-N-C催化剂的Fe含量(x轴)、*OH覆盖度(y轴)与OPM占比(z轴)的定量三维关联图。
要点四:工业级OER电催化性能
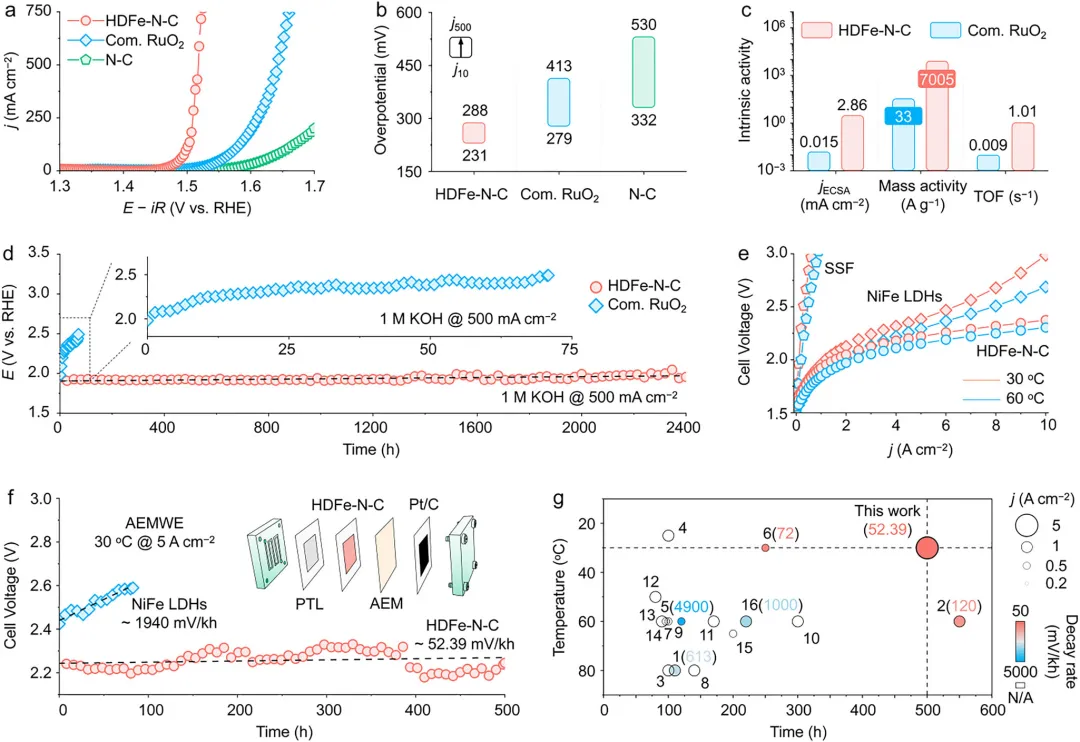
该催化剂在三电极体系和AEMWE器件中均展现出优异的工业级OER电催化性能,在1 M KOH 电解液的三电极体系中,HDFe-N-C在500 mA cm-1电流密度下过电位仅288 mV,远优于商业RuO2催化剂(413 mV),Tafel斜率低至40 mV dec-1,与OPM路径决速步(*O─*O 偶联)的理论值一致,且稳定性优异,在500 mA cm-1电流密度下连续运行2000 h,电压衰减率仅0.027 mV h-1,而商业RuO2在相同条件下仅能维持70 h;以HDFe-N-C为阳极组装的阴离子交换膜电解槽,在60℃下5 A cm-2工业级电流密度时电池电压仅2.15 V(能耗5.14 kWh Nm-3H2),优于NiFe LDH基准催化剂(2.29 V),30℃低温下5 A cm-2电流密度下可稳定运行500 h,性能衰减率比NiFe LDH低两个数量级,同时动态稳定性优异,经过35000次0~5 A cm-2快速启停循环后,电压衰减率仅2.74 mV kth-1,能够适配可再生能源间歇性供电的应用场景。
图4 HDFe-N-C催化剂的OER性能。(a) 三电极体系中HDFe-N-C、商业RuO2和N-C载体经95% iR校正后的OER极化曲线,以及HDFe-N-C快速循环伏安加速老化测试中活性衰减随扫描圈数的变化。(b) 分别在j10和j500电流密度下对应的过电位。(c) HDFe-N-C与商业RuO2的本征活性对比。(d) 不同电催化剂在 j500电流密度下未经iR校正的计时电位曲线。(e) HDFe-N-C与镍铁层状双氢氧化物(NiFe LDH)组装的阴离子交换膜水电解(AEMWE)池未经iR补偿的极化曲线。(f) HDFe-N-C与NiFe LDH阳极组装的AEMWE池在5 A cm-2恒电流密度下的计时电位曲线。(f) 的插图为AEMWE池结构示意图。(g) HDFe-N-C纳米片与近期报道的其他高性能AEMWE OER基准催化剂的稳定性对比。
本工作首次实现了Fe位点密度对Fe─N─C单原子催化剂OER反应路径的连续可控调控,通过制备超高负载的HDFe-N-C催化剂,成功触发了热力学更有利的OPM路径,突破了传统AEM的线性标度限制。该催化剂在工业级高电流密度下展现出突破性的活性和稳定性,为低成本、高效稳定的AEMWE析氧催化剂设计提供了全新策略,也为单原子催化剂的反应路径工程和工业化应用奠定了坚实基础。
Customized O─O Radical Coupling Route on High-Density Fe─N─C Catalysts for Stable Industrial-Scale Water Oxidation
https://doi.org/10.1002/anie.9504269
整理:付杰
编辑:张湘媛
欢迎关注我们,订阅更多最新消息
点击左下角“阅读原文”即可查看论文原文


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
