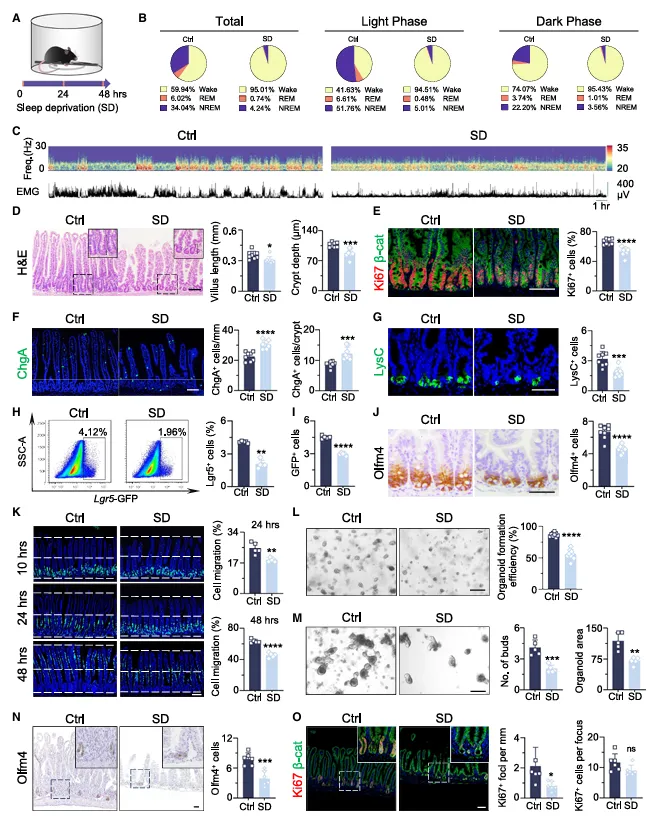
作者先确认小鼠确实没睡成觉——REM睡眠直接跌到0.74%,两天折腾下来,肠道隐窝和绒毛肉眼可见地缩水了。
潘氏细胞少了,但EC细胞反而多了。Lgr5和Olfm4阳性的干细胞明显减少,BrdU迁移变慢,类器官也长不好。说明缺觉直接把肠道的再生能力给削了。
我有点好奇,干细胞少了那么多,肠道屏障还能撑多久?
图1.急性睡眠剥夺损害肠道干细胞功能并破坏上皮细胞更新
想做睡眠剥夺对肠道的影响,别只看整体形态。把近端、中段、远端小肠分开分析,这篇文章发现近端损伤最重。
如果你用的模型不同,区域差异可能也不一样,建议先做一个区域梯度预实验,别一股脑全混着测。
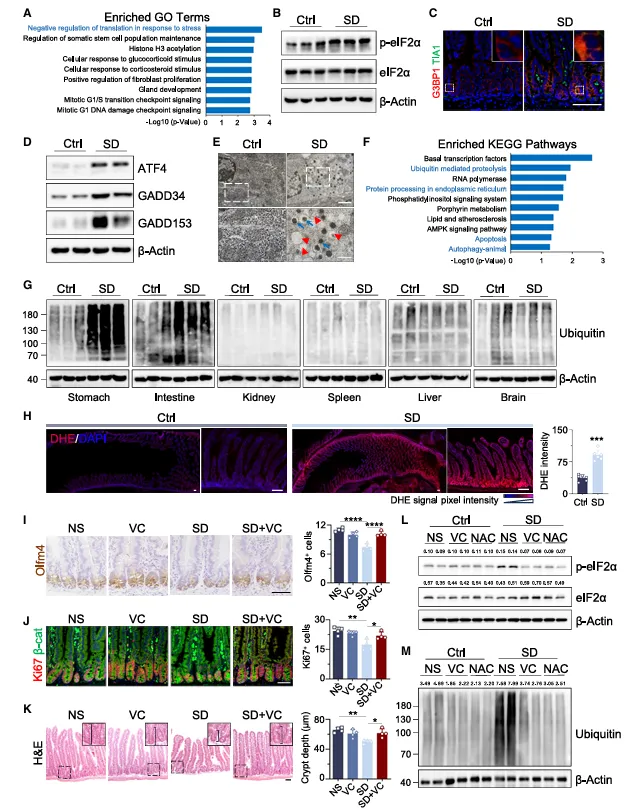
蛋白组学结果让我意外,最富集的通路居然是“应激下的翻译负调控”。p-eIF2α升高,应激颗粒增多,泛素化蛋白也堆起来了。翻译停了,错误折叠蛋白清不掉,干细胞等于被“噎死”了。
这条通路在其他应激里常见,但在缺觉导致的肠损伤里被证实,还是挺新颖的。
图2.短期睡眠剥夺通过激活翻译应激反应损害肠道干细胞
蛋白组学筛到了翻译应激通路,但他们没停在WB验证,而是用应激颗粒(TIA1和G3BP1)做功能验证。
建议你筛到某个应激通路后,别只做磷酸化检测,加一个应激颗粒染色,直观展示翻译是否真的停了。
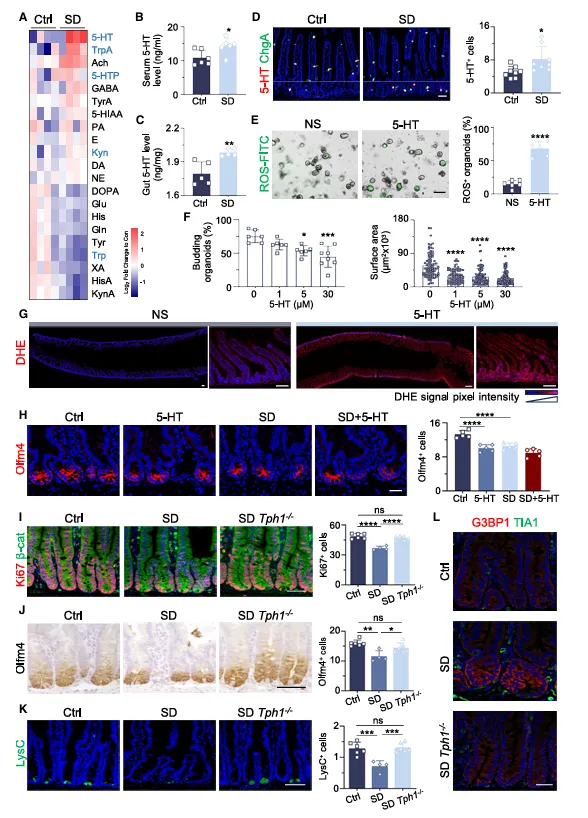
代谢组筛了一圈,肠道5-HT涨了近3倍,血清里的反而降了。DHE染色显示ROS很高,抗氧化剂能拉回来一点。Tph1敲除后,缺觉居然不伤肠了,隐窝深度和干细胞都正常。
这个遗传学证据锁定了EC细胞和5-HT,比前面药理实验更有说服力。
图3.急性睡眠剥夺破坏肠道5-HT稳态并诱导氧化应激
他们用Tph1全敲小鼠证明5-HT的来源是EC细胞,但全敲有发育代偿风险。
如果你有条件,建议做EC细胞特异性Tph1敲除,再复现一遍。这篇文章的结论需要更精细的遗传学验证来巩固。
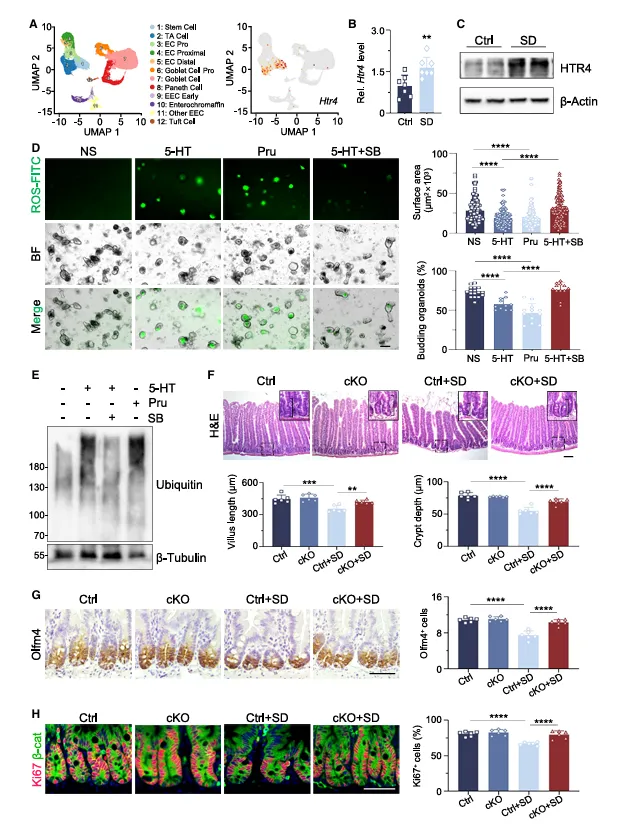
单细胞数据里,Htr4在干细胞中高表达,缺觉后受体本身还上调了。激动剂直接抑制类器官生长,拮抗剂能挽救。Htr4肠道敲除后,缺觉不伤肠了。
说明5-HT不是随便找个受体,而是精准靶向干细胞上的HTR4。这个“配体-受体”对应关系做得很干净。
图4.肠道干细胞通过其同源受体HTR4响应5-HT
验证受体功能时,他们同时用了激动剂(体外)和条件性敲除(体内)。
建议你也这样组合:体外用激动剂/拮抗剂快速筛表型,体内用条件性敲除确认必要性,两套证据互相支撑,让人挑不出毛病。
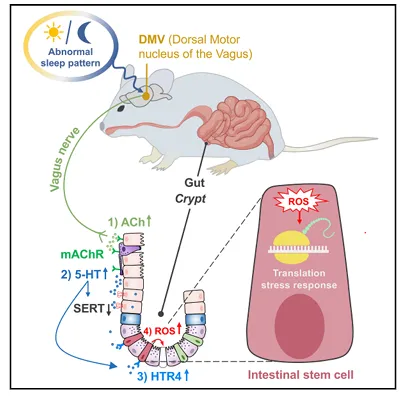
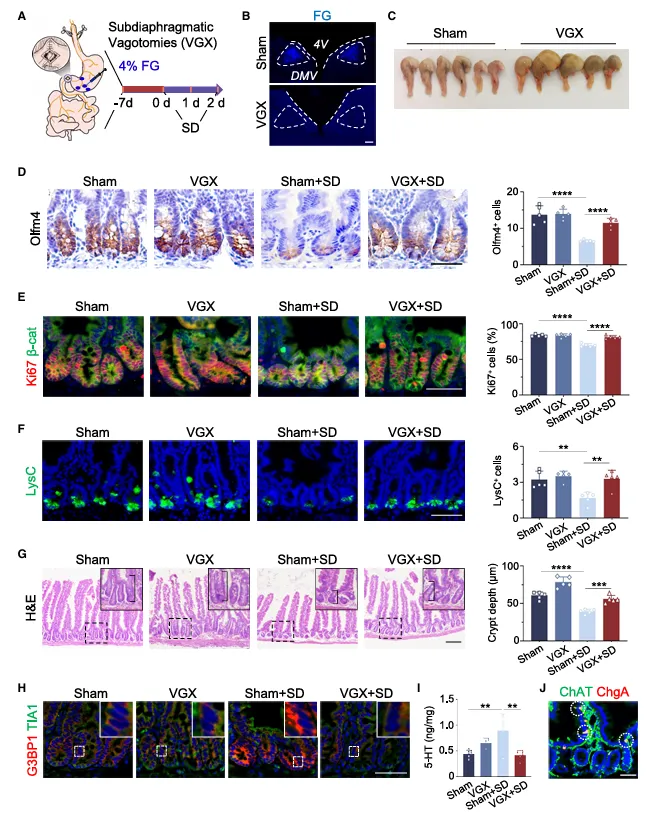
双侧迷走神经切断,缺觉的信号就传不下去了,肠道一切正常。5-HT也没升。这个手术实验有点粗糙但有效,粗暴地证明了脑-肠之间必须经过这条“电线”。
我注意到他们没做假手术对照组,但损伤和挽救的对比已经够说明问题了。
迷走神经切断术是个很“粗暴”的干预,但在这个场景里有效。
建议加一组假手术对照,虽然这篇文章没做,但你自己做的时候一定要加上,排除手术创伤对肠道的影响。
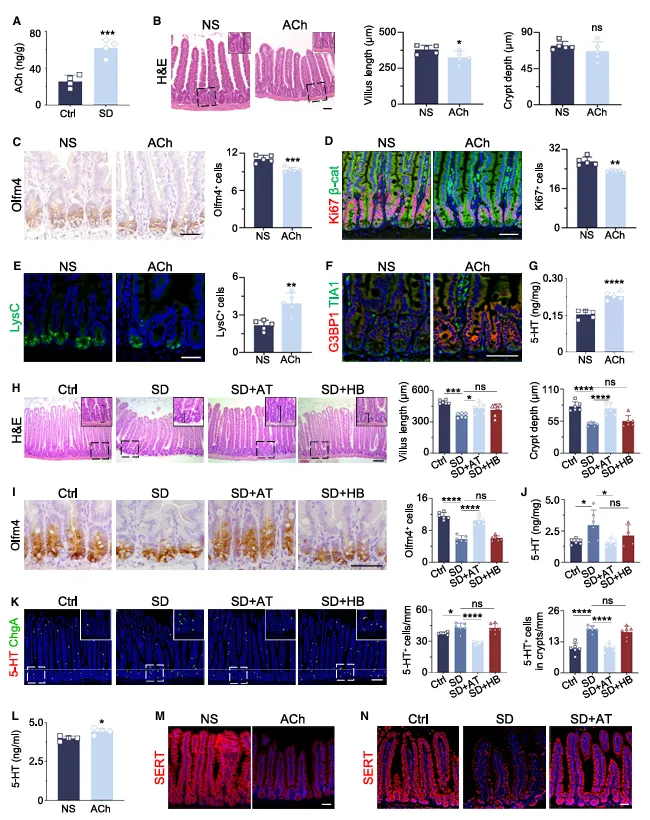
肠道Ach涨了近2倍,直接打Ach复现了所有表型。阿托品有效,烟碱拮抗剂无效,说明是毒蕈碱受体在干活。
更关键的是,Ach既促进EC细胞吐5-HT,又抑制SERT回收,一头产一头堵,5-HT不爆表才怪。这个“双向调控”的机制挺巧妙。
他们发现Ach既促进5-HT释放又抑制回收,一个配体两个作用。
如果你发现某个分子有类似的双向调控效应,建议分别阻断两个下游路径,看哪个占主导。这篇文章没拆这么细,你可以做得更深。
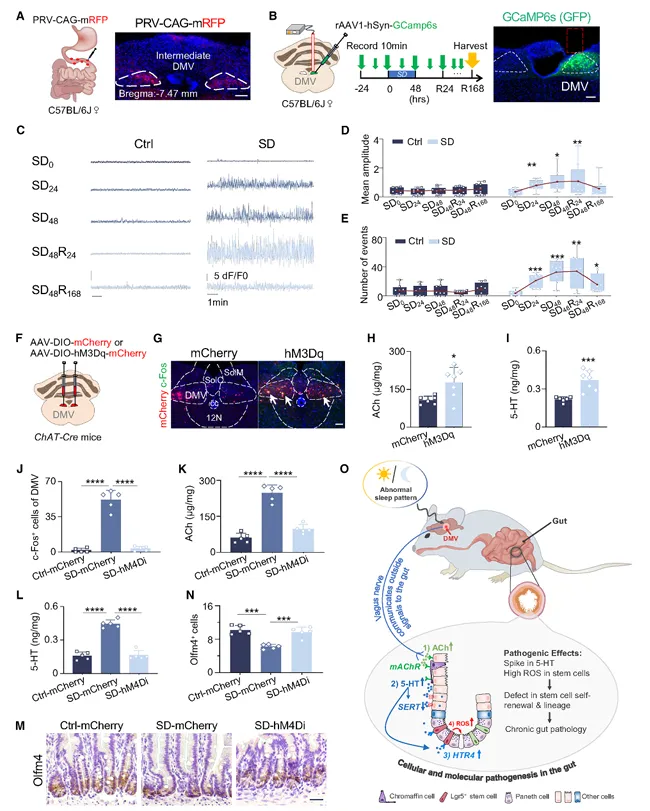
DMV的钙信号在缺觉后频率和幅度都蹿升了,而且高活动状态持续了7天。化学遗传激活DMV,Ach和5-HT都升高,干细胞减少。抑制DMV则阻断一切。DMV就是这个脑-肠通路的开关。
我好奇的是,长期失眠会不会让DMV一直处于这种高活动状态,导致肠道持续受损?
钙信号记录做了7天,发现DMV持续激活。建议你记录更长时间,看看慢性失眠模型里DMV会不会一直处于高活动状态。
这篇文章只做了急性缺觉,长期的影响还是未知的,值得追。













 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
