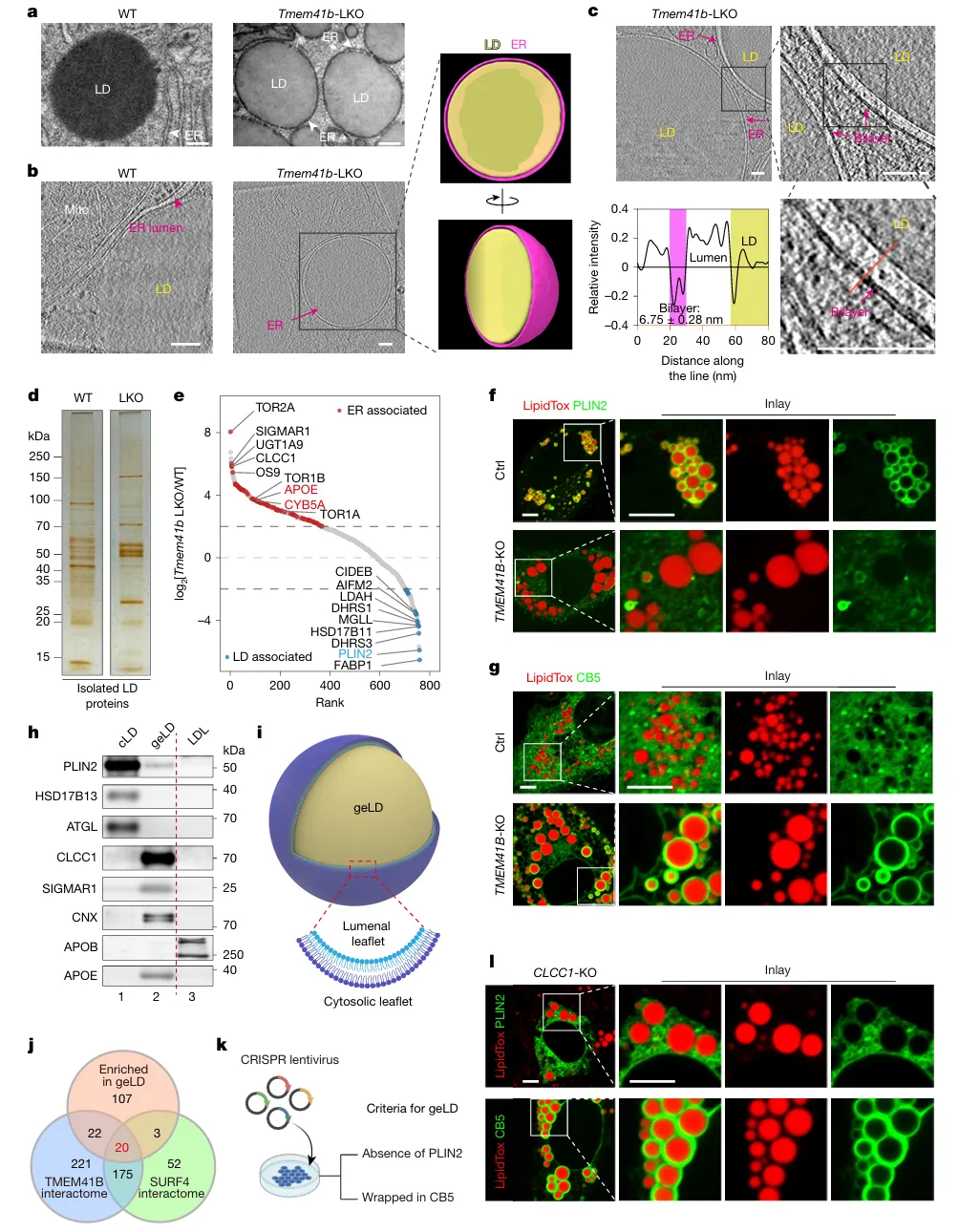
TMEM41B敲除肝细胞出现ER包裹的巨型脂滴(geLD),位于ER腔内,缺乏胞质脂滴标志PLIN2,富集ER膜蛋白。定量分析显示,geLD的磷脂总量仅为同等大小胞质脂滴的1.5倍(理论平衡值为3倍),直接证实ER双层磷脂分布不对称。多组学交集筛选锁定CLCC1,敲除CLCC1可完全复现geLD表型(图1)。
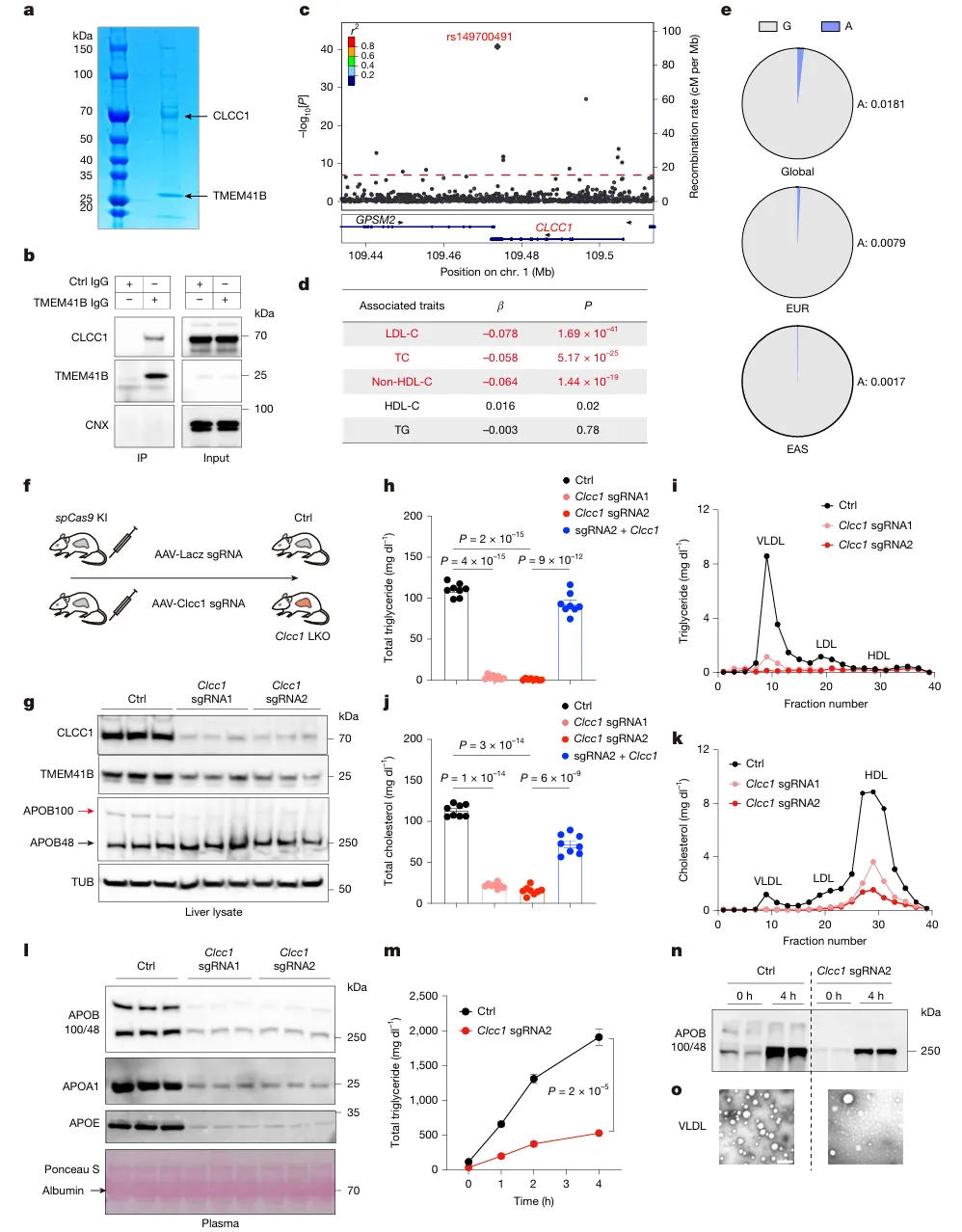
CLCC1与TMEM41B存在蛋白互作;肝脏敲除CLCC1会降低TMEM41B蛋白水平,但不影响其mRNA。人群GWAS数据显示,CLCC1遗传变异与血浆LDL-C、总胆固醇水平显著相关(P = 1.69 × 10⁻⁴¹)。小鼠肝脏敲除CLCC1后,血浆甘油三酯、胆固醇接近清零,VLDL分泌几乎丧失;回补CLCC1可完全逆转(图2)。
图2.CLCC1与TMEM41B共同控制脂质批量输出
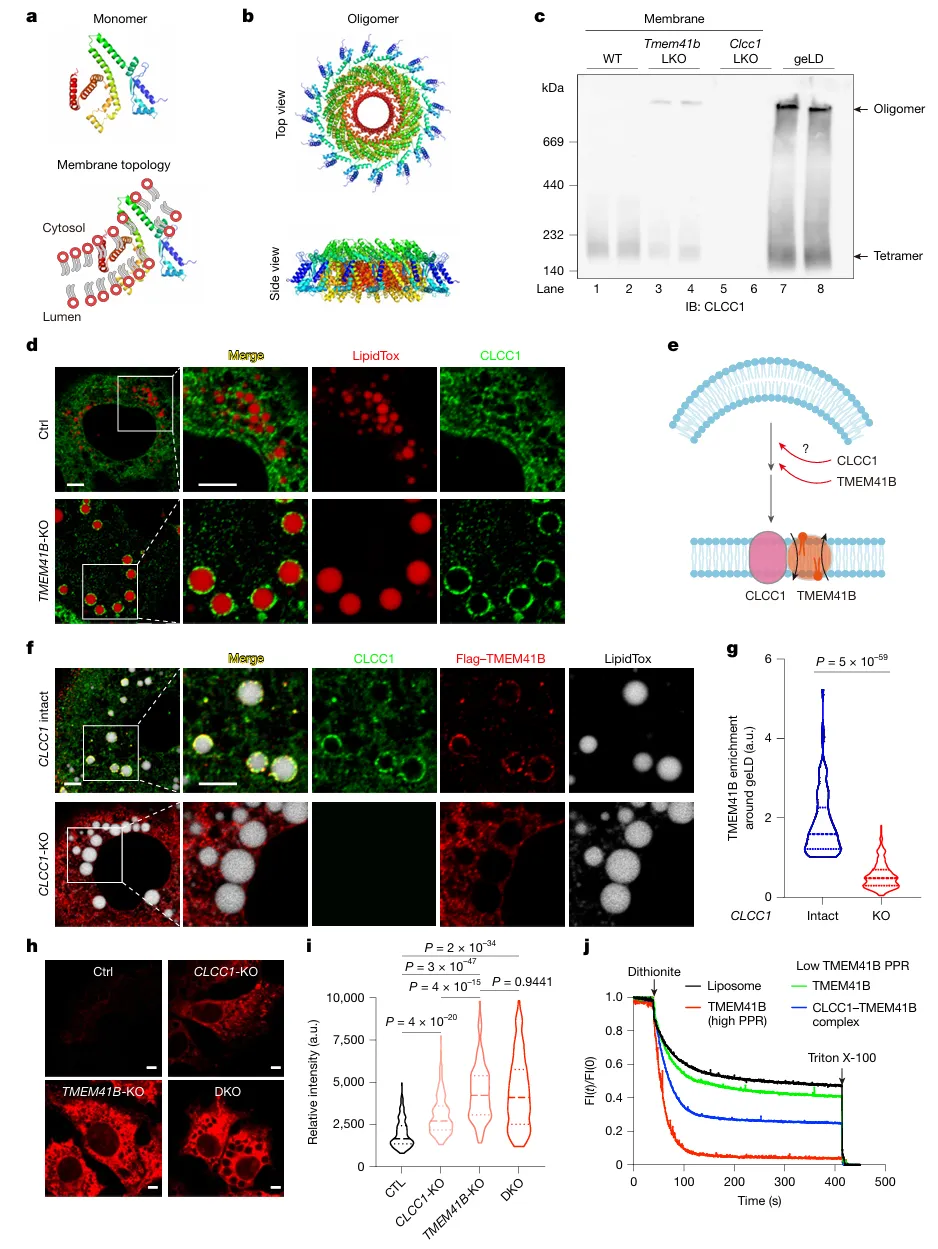
CLCC1可形成环状多聚体;磷脂失衡时CLCC1发生寡聚化,富集到geLD周围的ER膜,其腔侧结构域是寡聚化与定位的必要条件。CLCC1是TMEM41B定位到失衡ER膜的必需条件。CLCC1自身无翻转酶活性,但可显著增强TMEM41B的磷脂翻转效率——体外实验显示,CLCC1-TMEM41B复合体的翻转活性约为等量TMEM41B单独作用的2倍以上(图3)。
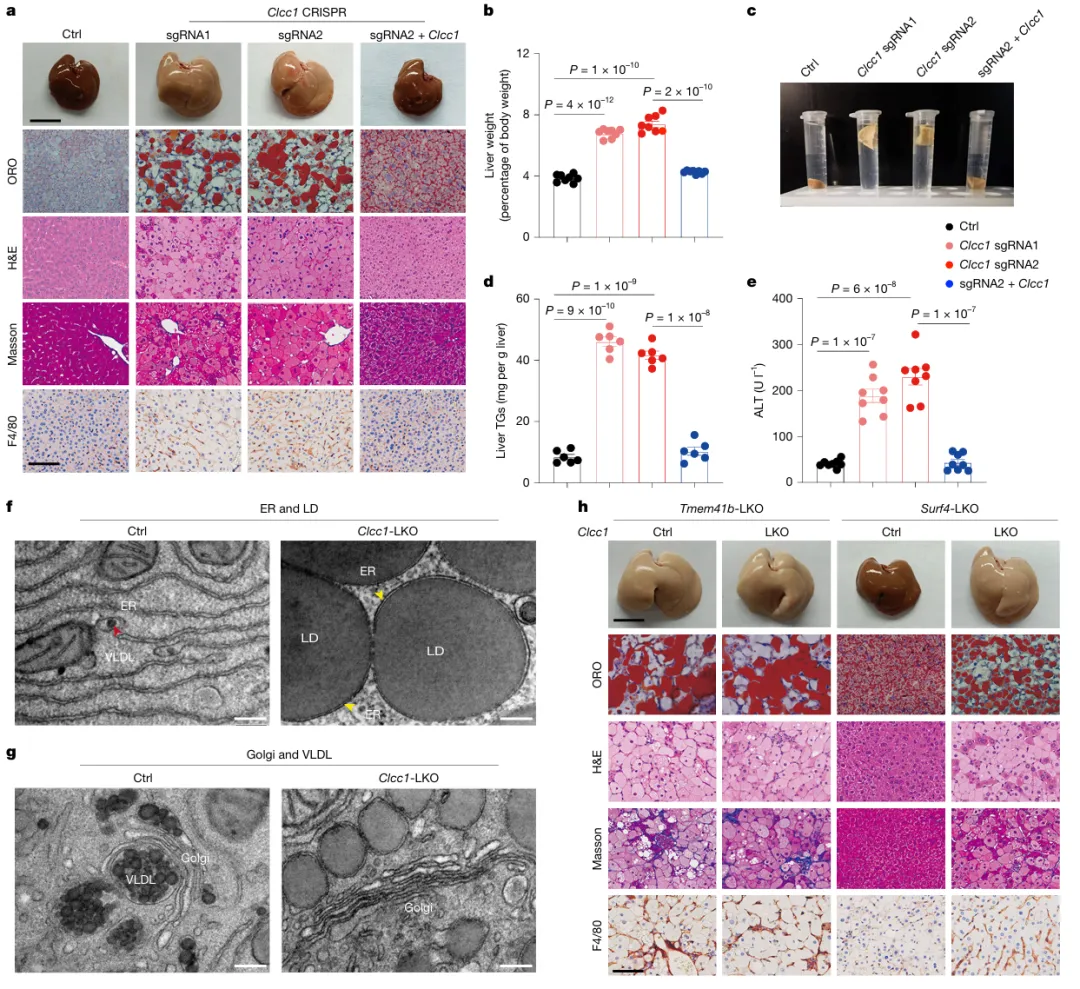
小鼠肝脏敲除CLCC1仅4周,无需高脂饮食即可自发出现典型MASH表型,回补CLCC1可逆转。超微结构显示,敲除小鼠肝细胞ER中大量geLD堆积,高尔基体中完全无VLDL颗粒。上位性实验证实:CLCC1与TMEM41B同处一条通路,作用于脂蛋白组装阶段,早于SURF4介导的运输步骤(图4)。
图4.CLCC1缺失加速代谢功能障碍相关脂肪性肝炎进展
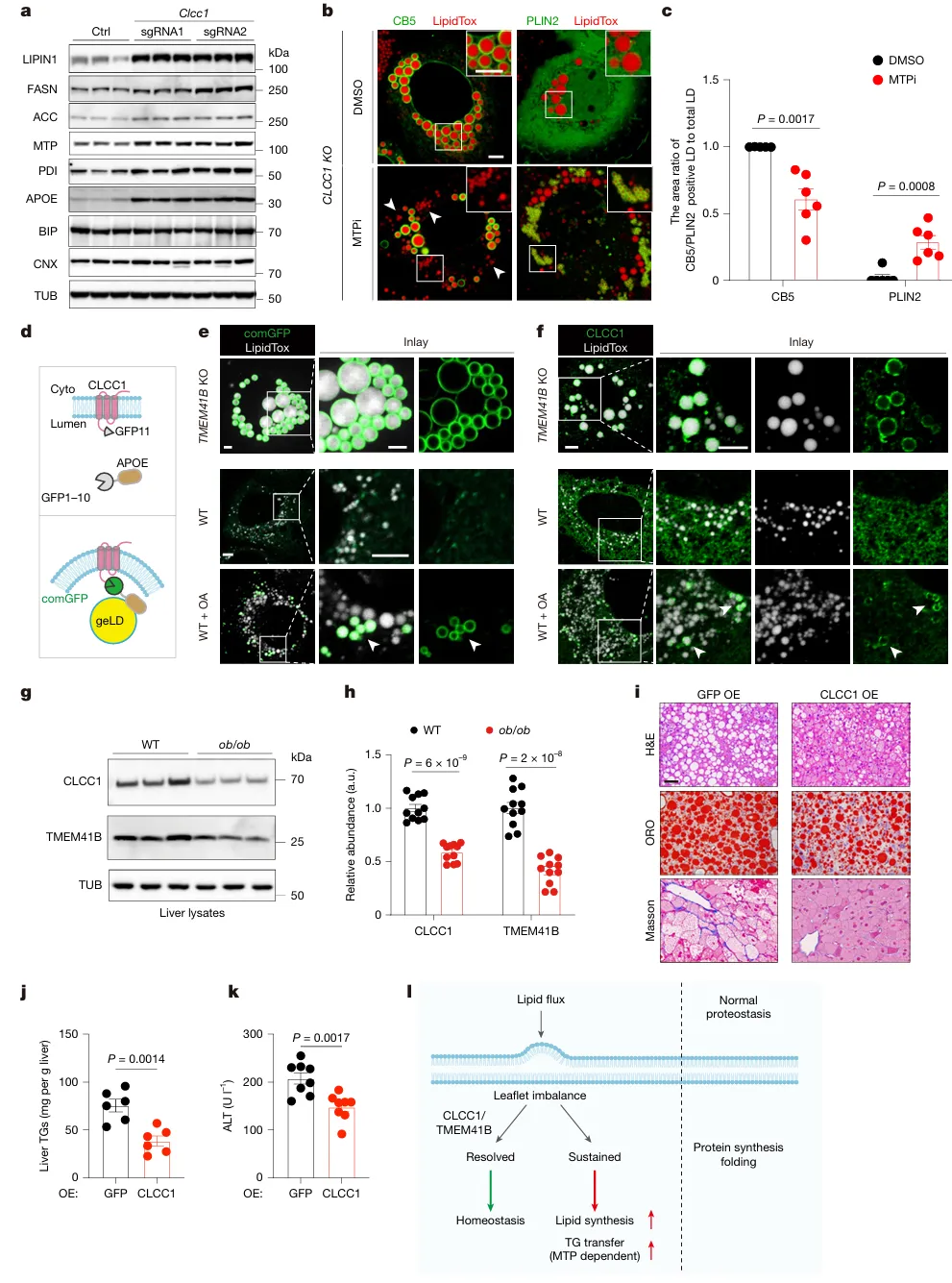
CLCC1缺失会上调脂合成酶与MTP作为代偿;抑制MTP可将geLD重新转变为胞质脂滴。GFP互补报告系统证实,生理脂质负荷增加也会引发ER磷脂失衡,诱导CLCC1响应。肥胖小鼠肝脏CLCC1/TMEM41B蛋白下调;肝脏过表达CLCC1可显著减轻肝脂堆积与肝损伤(图5)。
















 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
