作者
王浩(郑州大学)
卢思宇(郑州大学)
引言
1. 酸性析氧反应(OER)作为质子交换膜水电解槽(PEMWEs)的阳极关键反应,受限于固有的迟缓四电子转移和O=O键形成动力学,严重制约电解水制氢效率。目前商用的IrO₂基催化剂虽性能优异,但Ir储量极低且成本高昂(约140美元/克),难以支撑PEMWEs的大规模商业化。
2. RuO₂因理论活性优于IrO₂且价格相对低廉,被视为替代IrO₂的首选阳极材料。然而在酸性高电位(>1.4 V vs. RHE)环境下,RuO₂基材料易发生结构坍塌与溶解,存在固有的活性-稳定性权衡难题。
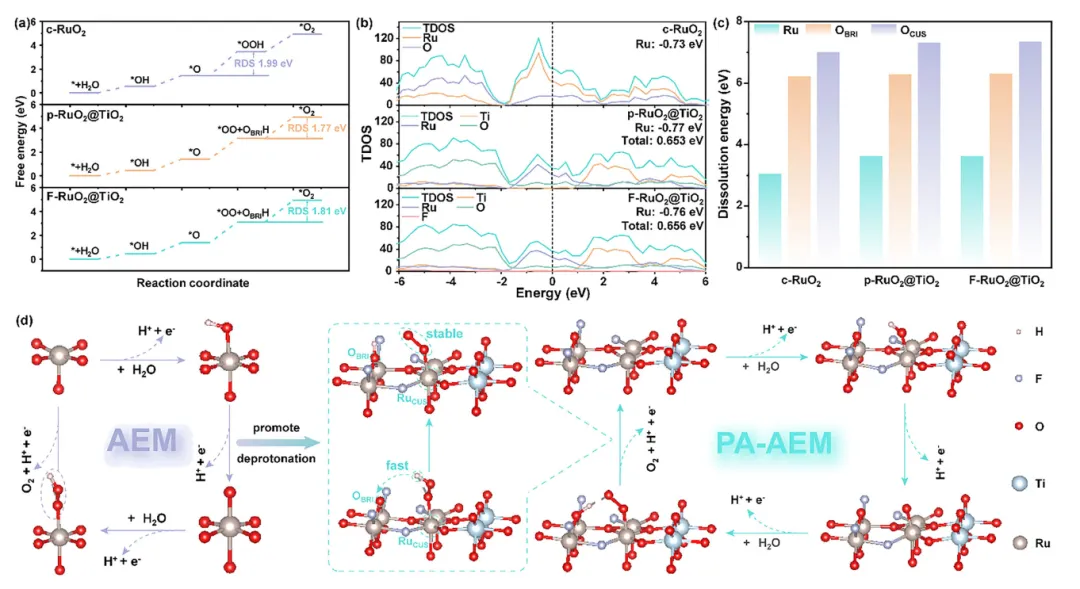
3. 传统吸附演化机制(AEM)受*OH与*OOH吸附能标度关系制约,且高质子浓度下*OOH中间体的O-H键断裂受阻;晶格氧介导机制(LOM)虽能打破标度关系,但晶格氧的逃逸速率远超再生速率,导致催化剂结构迅速崩塌。
4. 因此,亟需开发既能促进质子转移、又能维持结构完整性的新策略,以同步提升RuO₂基催化剂在酸性OER中的活性与稳定性。
核心发现
1. 通过混合喷雾退火法合成了F掺杂RuO₂负载于TiO₂的电催化剂(F-RuO₂@TiO₂),构建了Ru-O-Ti界面平台实现电负性介导的电荷重分布。NH₄X在热分解过程中释放HX,对Ti基底产生温和刻蚀作用,促进卤素掺杂RuO₂在原位生成的TiO₂上均匀负载。
2. F-RuO₂@TiO₂在0.5 M H₂SO₄中展现出优异的OER活性,过电位仅为170、207、224和236 mV(分别在10、50、100和200 mA cm⁻²下),显著优于c-RuO₂(η₁₀=258 mV)。其Tafel斜率最小,表明反应动力学更有利。
3. 该催化剂具有卓越的长期稳定性,在100和200 mA cm⁻²下分别稳定运行660 h和253 h,法拉第效率约99.5%,S-number约为9.5×10⁴(是c-RuO₂的15.7倍),有效抑制Ru溶解。
4. 组装的PEMWE在80°C下仅需1.57和1.68 V即可达到0.5和1 A cm⁻²,并在60°C下分别稳定运行300和100 h,远超c-RuO₂(1 A cm⁻²下约2 h完全失活)。
5. 机理研究表明,F的高电负性增强了Ru-O共价性,通过质子辅助吸附演化机制(PA-AEM)加速*OOH去质子化;同时Ru-O-Ti间的动态电荷重分布使TiO₂缓冲Ru位点的电荷波动,有效抑制过氧化。DFT计算证实TiO₂主要提升活性(d带中心从-0.73降至-0.77 eV),F主要增强稳定性(提高Ru和O_L的溶解能)。
图文解读
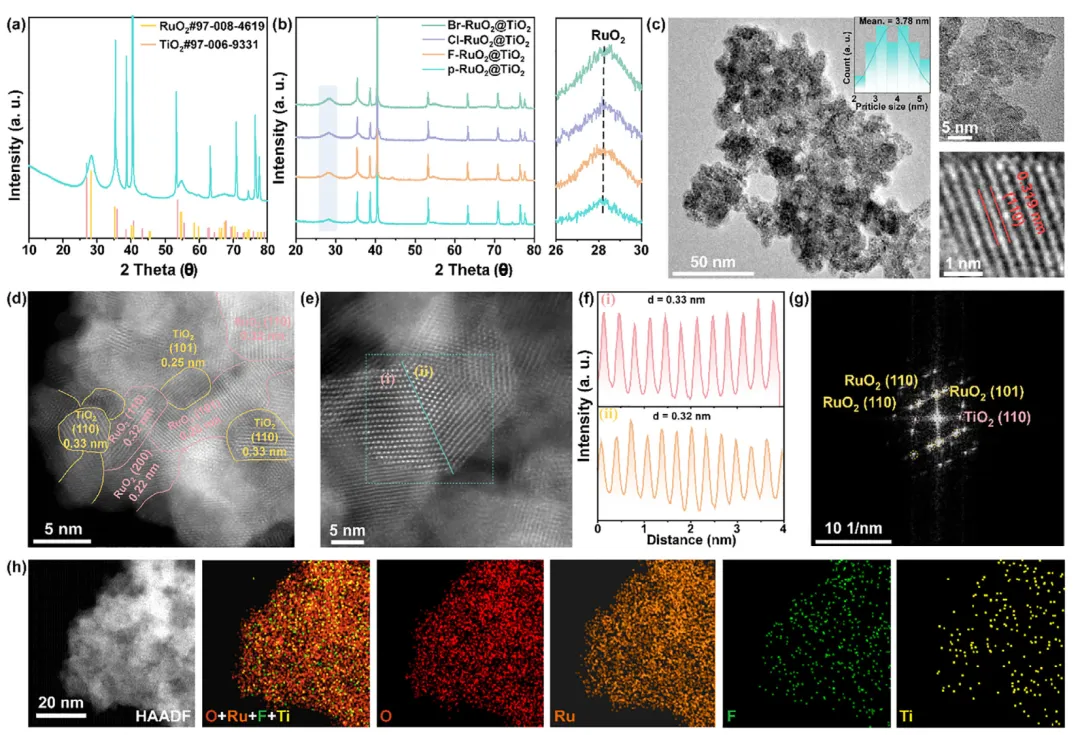
图1:X-RuO₂@TiO₂的晶体结构与微观形貌表征
1. 成功构建RuO₂/TiO₂异相界面:慢速XRD扫描显示Ti基底在退火过程中部分氧化为金红石TiO₂(PDF#97-006-9331),与RuO₂同属四方晶系(空间群P42/mnm),HAADF-STEM证实0.33 nm的TiO₂(110)晶面与0.32 nm的RuO₂(110)晶面紧密耦合,FFT图案进一步确认两相共存。 2. 卤素掺杂引起晶格收缩:相比p-RuO₂@TiO₂的0.325 nm晶面间距,F/Cl/Br掺杂后RuO₂(110)面间距缩小至约0.32 nm,XRD衍射峰向高2θ方向偏移,表明掺杂导致晶格收缩。TEM-EDX元素映射证实卤素原子成功掺入且RuO₂晶粒明显细化,有利于增大电化学活性面积。 3. 掺杂样品形成连续多孔网络:SEM显示卤素掺杂的RuO₂@TiO₂在Ti基底上形成连续互连的多孔网络结构,均匀覆盖Ti纤维并暴露丰富活性位点;而未掺杂的p-RuO₂@TiO₂分布不连续,界面集成度较弱。
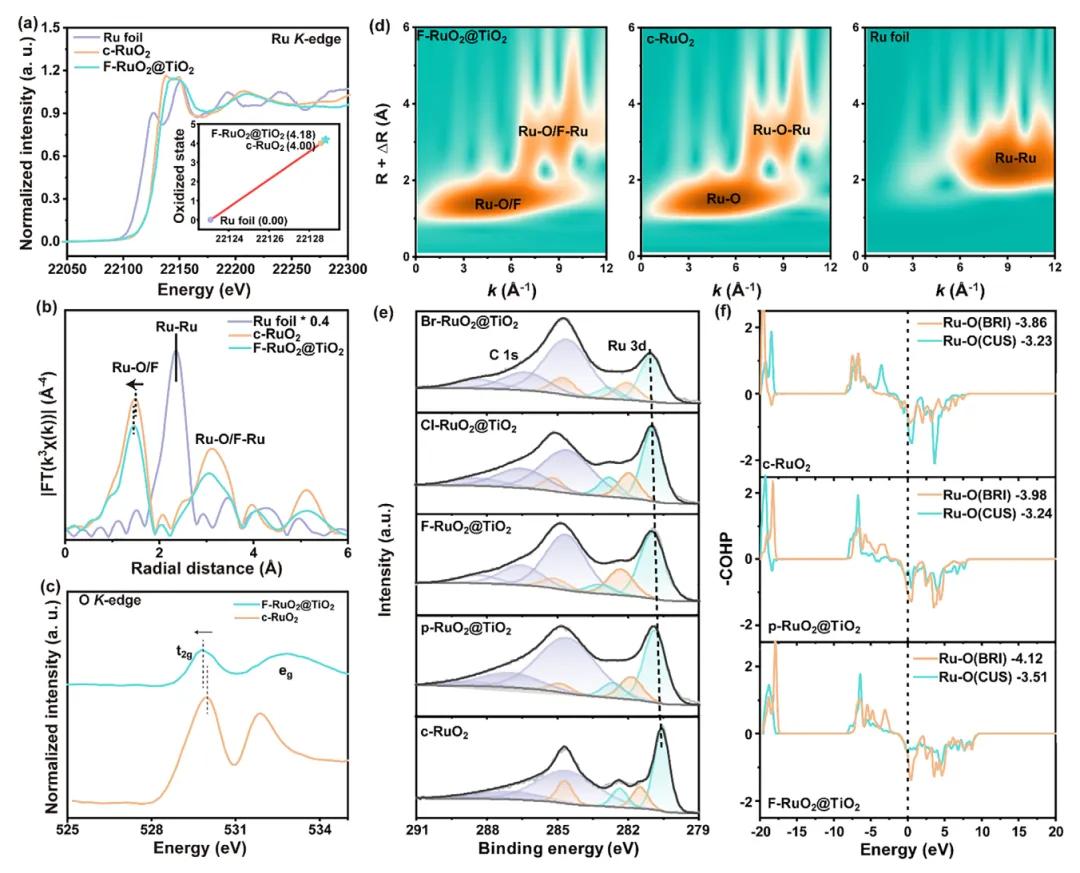
图2:X-RuO₂@TiO₂的电子结构与价态分析
1. F掺杂提升Ru价态并增强Ru-O共价性:Ru K-edge XANES显示F-RuO₂@TiO₂吸收边明显右移,表明Ru氧化态升高;EXAFS拟合显示Ru-O/F键长从1.99 Å缩短至1.96 Å,COHP计算证实Ru-O键的绝对值在CUS和BRI位点均为最高,表明F掺杂显著增强Ru-O共价性。 2. TiO₂支撑层驱动动态电荷补偿:XPS显示Ti 4p结合能降低0.1 eV,表明Ti得到电子;Ru 3d结合能升高,表明Ru失去电子。这种反向电荷补偿机制使TiO₂向缺电子Ru中心转移电子,抑制Ru的进一步氧化,氧空位(O_V)的缺失进一步证实了该动态电荷补充机制。 3. F的电负性诱导位点依赖性电荷极化:Bader电荷分析显示,强电负性的F主要影响桥位Ru原子(RuBRI),提升其氧化态;而配位不饱和Ru原子(RuCUS)作为电子储库出现轻微电子积累,形成位点依赖的电荷极化,整体上Ru平均氧化态略有升高,与XPS结果一致。
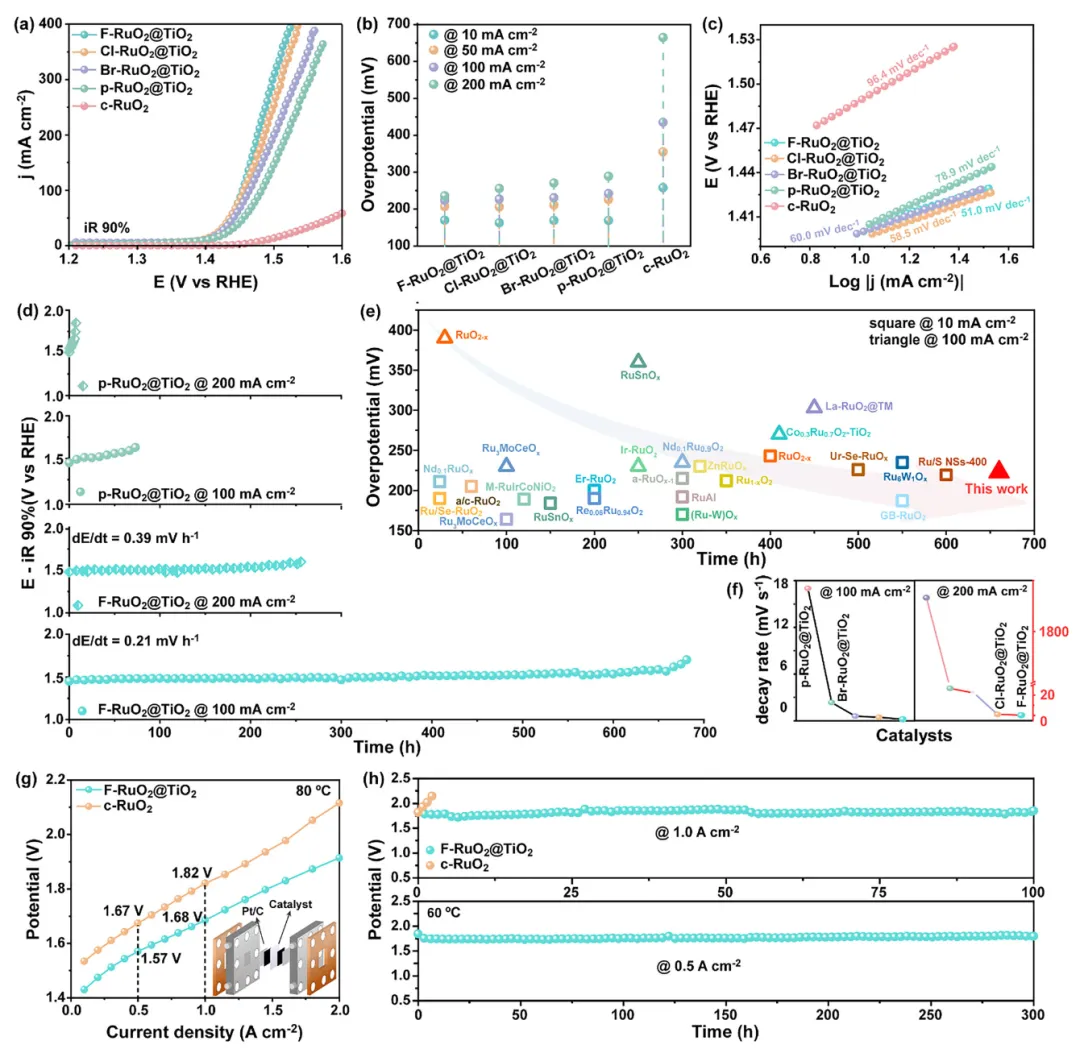
图3:X-RuO₂@TiO₂的酸性OER性能与PEMWE表现
1. F-RuO₂@TiO₂在高电流密度下展现最低过电位:在100和200 mA cm⁻²下过电位仅224和236 mV,优于Cl-RuO₂@TiO₂(227/256 mV)、Br-RuO₂@TiO₂(231/271 mV)、p-RuO₂@TiO₂(244/289 mV)和c-RuO₂(435/665 mV)。F的最佳掺杂含量为3.45 at%,活性-掺杂量呈火山型关系。 2. 长期稳定性远超对比样品:F-RuO₂@TiO₂在100和200 mA cm⁻²下分别稳定运行660 h和250 h,电压衰减率极低;而Cl/Br-RuO₂@TiO₂降解明显加快,p-RuO₂@TiO₂和c-RuO₂电位急剧升高。S-number约9.5×10⁴,是c-RuO₂的15.7倍,证实其抗Ru溶解能力。 3. PEMWE器件性能优异:以F-RuO₂@TiO₂为阳极、Pt/C为阴极、Nafion 115为质子交换膜组装的电解槽,在80°C下仅需1.57和1.68 V即可达到0.5和1 A cm⁻²(c-RuO₂需1.80 V@1 A cm⁻²),并在60°C下分别稳定运行300和100 h,而c-RuO₂在1 A cm⁻²下约2 h完全失活。
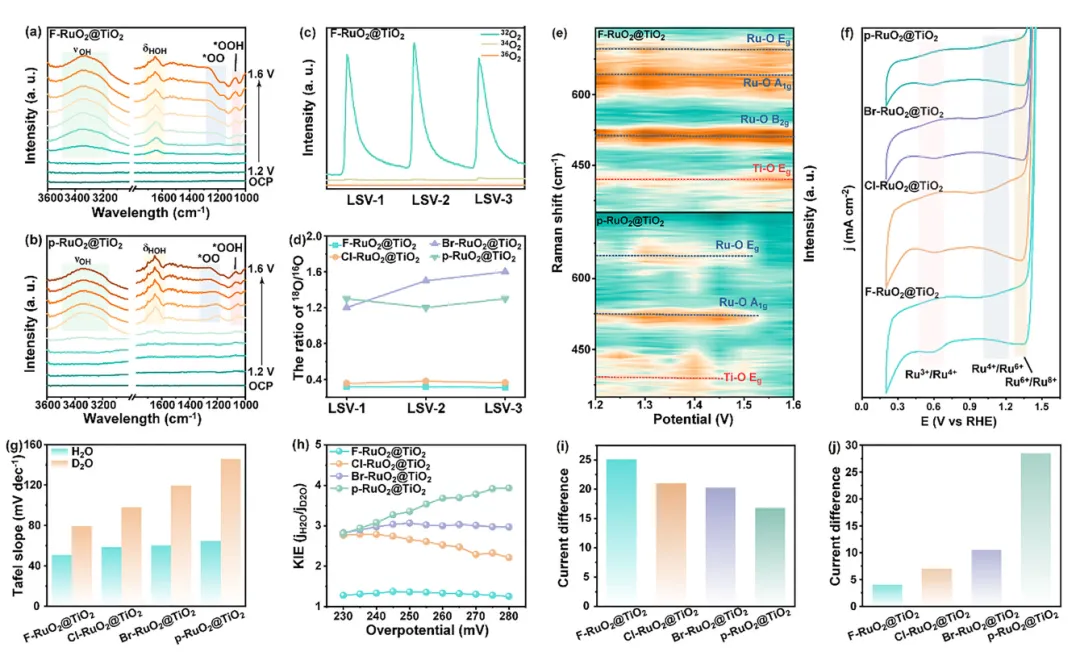
图4:OER机理的原位表征与电化学分析
1. PA-AEM路径抑制晶格氧参与:原位ER-IR在~1060 cm⁻¹和~1200 cm⁻¹检测到*OOH和*OO中间体共存,证实*OOH去质子化过程。DEMS的¹⁸O同位素标记实验显示F-RuO₂@TiO₂的¹⁸O产物比例接近自然丰度(~0.2%),表明*OO中间体主要来源于电解液而非晶格氧,有效抑制O_L参与;而Cl/Br/p-RuO₂@TiO₂的¹⁸O比例显著偏高,说明O_L被过度消耗。 2. F掺杂稳定RuO₂骨架并抑制高价位态氧化:原位Raman显示F-RuO₂@TiO₂的Ti-O和Ru-O特征峰在宽电位范围内保持不变,而p-RuO₂@TiO₂的特征峰在1.5 V vs. RHE时显著减弱消失并发生蓝移。CV测试中仅F-RuO₂@TiO₂未出现Ru⁴⁺/Ru⁶⁺和Ru⁶⁺/Ru⁸⁺氧化还原峰,证实F有效抑制Ru向不稳定高价位态转化。 3. 电负性调控质子转移动力学:KIE测试显示Tafel斜率和KIE值按F-RuO₂@TiO₂ < Cl-RuO₂@TiO₂ < Br-RuO₂@TiO₂ < p-RuO₂@TiO₂顺序递增,直接证明高电负性阴离子通过激活相邻O_BRI作为质子受体,促进*OOH去质子化形成*OO和*H。TMA⁺探针实验进一步证实F-RuO₂@TiO₂对*OO中间体的稳定能力最强。
图5:DFT计算揭示AEM到PA-AEM路径转变机制
1. PA-AEM路径降低速控步能垒:c-RuO₂遵循传统AEM机制,速控步为*O形成*OOH步骤;而p-RuO₂@TiO₂和F-RuO₂@TiO₂在PA-AEM路径下速控步转变为*OOH中间体的去除,所需能垒显著降低。TiO₂促进H从*OOH转移到O_BRI位点形成*OO和O_BRIH,加速*OOH去质子化。 2. TiO₂与F的分工协同优化性能:TDOS分析表明TiO₂主要提升活性,使Ru的d带中心从-0.73 eV移至-0.77 eV,增强Ru-Ti电子耦合;F主要增强稳定性,增加Ru的d轨道与O的p轨道重叠,使Ru-O共价键更稳定。两者协同实现活性与稳定性的平衡。 3. F显著提升Ru和晶格氧的溶解能:溶解能计算清晰显示F的引入大幅提高Ru原子和O_L(在CUS和BRI位点)的溶解能,从热力学角度证实F能够稳定O_L并抑制Ru溶解,从而增强催化剂稳定性。基于此提出PA-AEM路径可同时优化传统AEM的活性和LOM的稳定性。
总结
本研究通过混合喷雾退火法合成F掺杂RuO₂负载于TiO₂的电催化剂,利用F的高电负性与TiO₂的支撑作用构建Ru-O-Ti界面平台,实现电负性介导的电荷重分布。F-RuO₂@TiO₂在酸性OER中展现出低过电位(224 mV@100 mA cm⁻²)和卓越稳定性(660 h@100 mA cm⁻²),组装的PEMWE在1.57 V下达到0.5 A cm⁻²并稳定运行300 h。机理研究证实F激活相邻O_BRI作为质子受体,通过PA-AEM路径加速*OOH去质子化并抑制晶格氧参与;TiO₂则通过动态电荷补偿缓冲Ru位点电荷波动,抑制过氧化。该工作建立了电负性调控质子转移动力学稳定RuO₂的新策略,为理性设计高性能RuO₂基酸性OER电催化剂提供了重要指导。
原文链接
https://doi.org/10.1002/anie.9750153


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
