郑州大学附属肿瘤医院 | 揭秘 USP25 在头颈鳞癌中的关键作用:逆转免疫抑制微环境,为免疫治疗添新靶点
- 2026-06-24 15:16:51

概述
为探究去泛素化酶在头颈鳞癌(HNSCC)肿瘤免疫微环境(TIME)中的作用,通过生物信息学筛选差异表达去泛素化酶,发现 USP25 在 HNSCC 中低表达且与不良预后相关;进一步结合单细胞测序、体内外功能实验及机制研究,证实 USP25 通过 UIM2 结构域结合 TAB2 并去除其 K63 连接泛素链,抑制 MAPK 通路激活及 IL-6 分泌,从而减少髓系来源抑制细胞(MDSCs)迁移、促进 CD8+T 细胞浸润,逆转免疫抑制微环境;最终发现 USP25 过表达可增强 HNSCC 对 PD-1 抑制剂的敏感性,为 HNSCC 免疫治疗提供新靶点。
注:该文章发表于《Cell Death Discovery》,该期刊最新影响因子为7.0,JCR分区Q1,中科院生物学大类2区。
研究要点解析
首次揭示 USP25 在 HNSCC 免疫微环境中的调控作用,明确其通过结合 TAB2 去除 K63 泛素链抑制 MAPK-IL-6 通路,调控 MDSCs 与 CD8+T 细胞浸润平衡;证实 USP25 低表达与 HNSCC 不良预后相关,且过表达可增强抗 PD-1 治疗敏感性,为 HNSCC 预后评估及免疫治疗联合靶点开发提供全新思路。
采用多组学生物信息学分析,从 TCGA、GEO 数据库筛选 HNSCC 与正常组织中差异表达的去泛素化酶,通过 GEPIA2、Kaplan-Meier Plotter 分析 USP25 的预后价值;构建 4-NQO 诱导的 HNSCC 小鼠模型及 MOC1 同源肿瘤模型,结合 USP25 过表达 / 敲低细胞系,通过 CCK8、Transwell、伤口愈合实验检测细胞功能;利用单细胞 RNA 测序(scRNA-seq)解析肿瘤免疫微环境细胞组成;通过免疫沉淀(Co-IP)、免疫荧光共定位、泛素化实验验证 USP25 与 TAB2 的相互作用及去泛素化机制;采用 IHC、mIHC 检测组织中 USP25、CD8+T 细胞、MDSCs 浸润水平;通过体内抗 PD-1 治疗实验评估 USP25 对免疫治疗疗效的影响;运用 ELISA、Western blot 检测细胞因子分泌及通路蛋白表达。
生物信息学分析显示,USP25 是 HNSCC 中唯一与不良预后相关的低表达去泛素化酶,其表达水平与 CD8+T 细胞浸润正相关、与 MDSCs 浸润负相关;体内实验证实 USP25 低表达促进 HNSCC 进展及淋巴结转移,过表达则缩小肿瘤体积;单细胞测序表明 USP25 低表达组存在免疫抑制性微环境,CD8+T 细胞比例降低、MDSCs 积累;机制研究发现 USP25 通过 UIM2 结构域结合 TAB2,去除其 K63 连接泛素链,抑制 MAPK 通路激活及 AP-1 介导的 IL-6 分泌,进而减少 MDSCs 迁移;功能实验显示 USP25 过表达可增强 T 细胞增殖及 IFN-γ 分泌,减弱 MDSCs 的免疫抑制作用;体内抗 PD-1 治疗实验证实,USP25 过表达组肿瘤对 PD-1 抑制剂敏感性显著提高,肿瘤内功能型 CD8+T 细胞比例增加、MDSCs 积累减少。
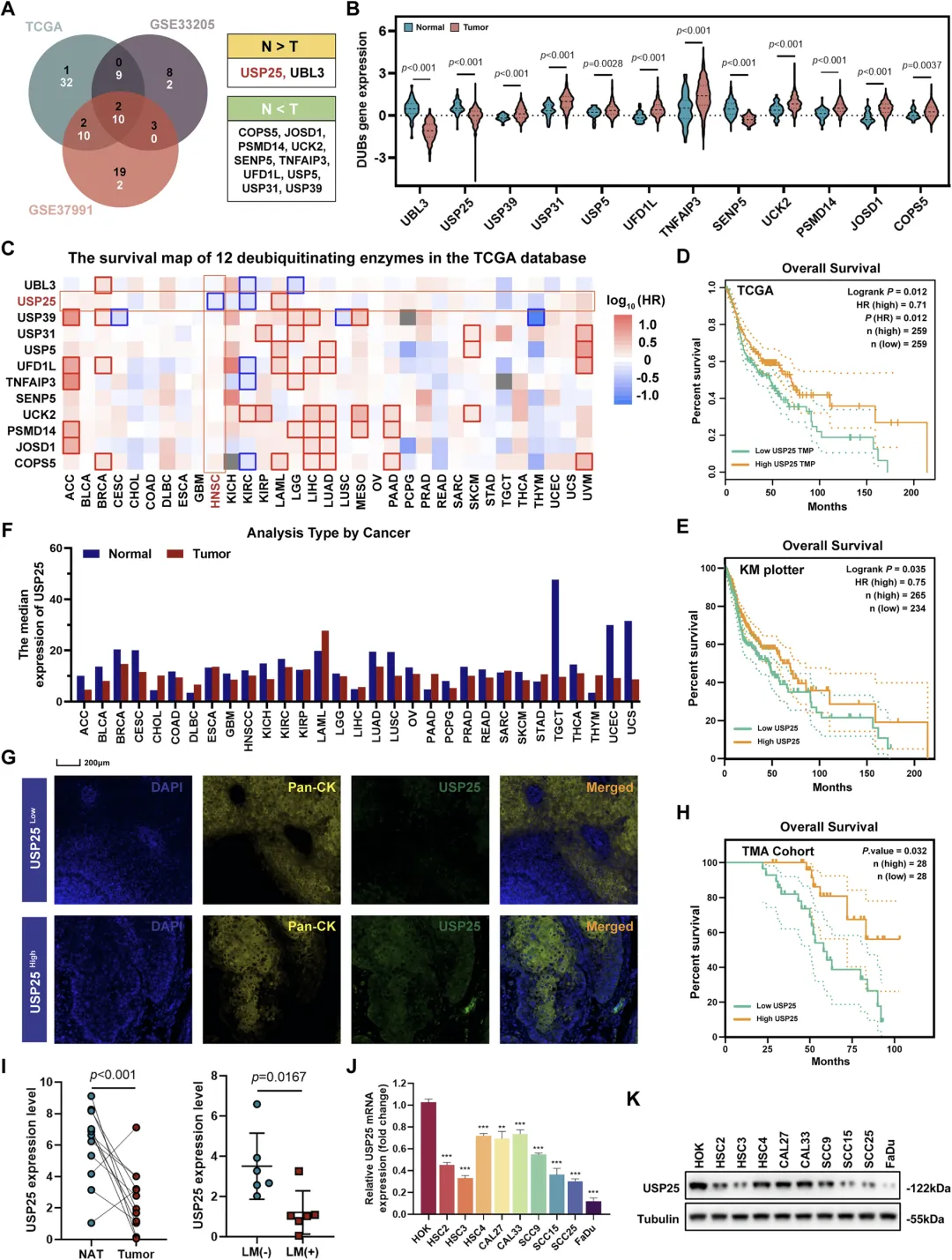
Figure 1:USP25 在 HNSCC 中低表达且与不良预后相关
该图通过多维度分析明确了 USP25 在 HNSCC 中的表达特征及临床意义。首先,通过 Venn 图和小提琴图从 TCGA、GEO 等数据库的 95 种去泛素化酶中,筛选出 HNSCC 与正常组织中 2 种显著下调(USP25、UBL3)和 10 种显著上调的去泛素化酶;随后经 GEPIA2、Kaplan-Meier Plotter 等工具分析,证实 USP25 是唯一与 HNSCC 患者总生存期缩短相关的去泛素化酶,低表达 USP25 的患者临床结局更差。同时,TCGA 数据库分析显示 USP25 在包括 HNSCC 在内的多种癌症中均呈下调趋势;组织芯片(TMA)的 mIHC 染色及生存分析进一步验证了这一结论。此外,qRT-PCR 结果表明 HNSCC 组织中 USP25 表达较癌旁正常组织显著降低,且淋巴结转移阳性的肿瘤组织中 USP25 表达更低;qPCR 和 Western blot 实验则证实,与正常人口腔角质形成细胞(HOK)相比,所有 HNSCC 细胞系中 USP25 的 mRNA 和蛋白水平均明显下调,综合表明 USP25 低表达与 HNSCC 恶性进展及不良预后密切相关。
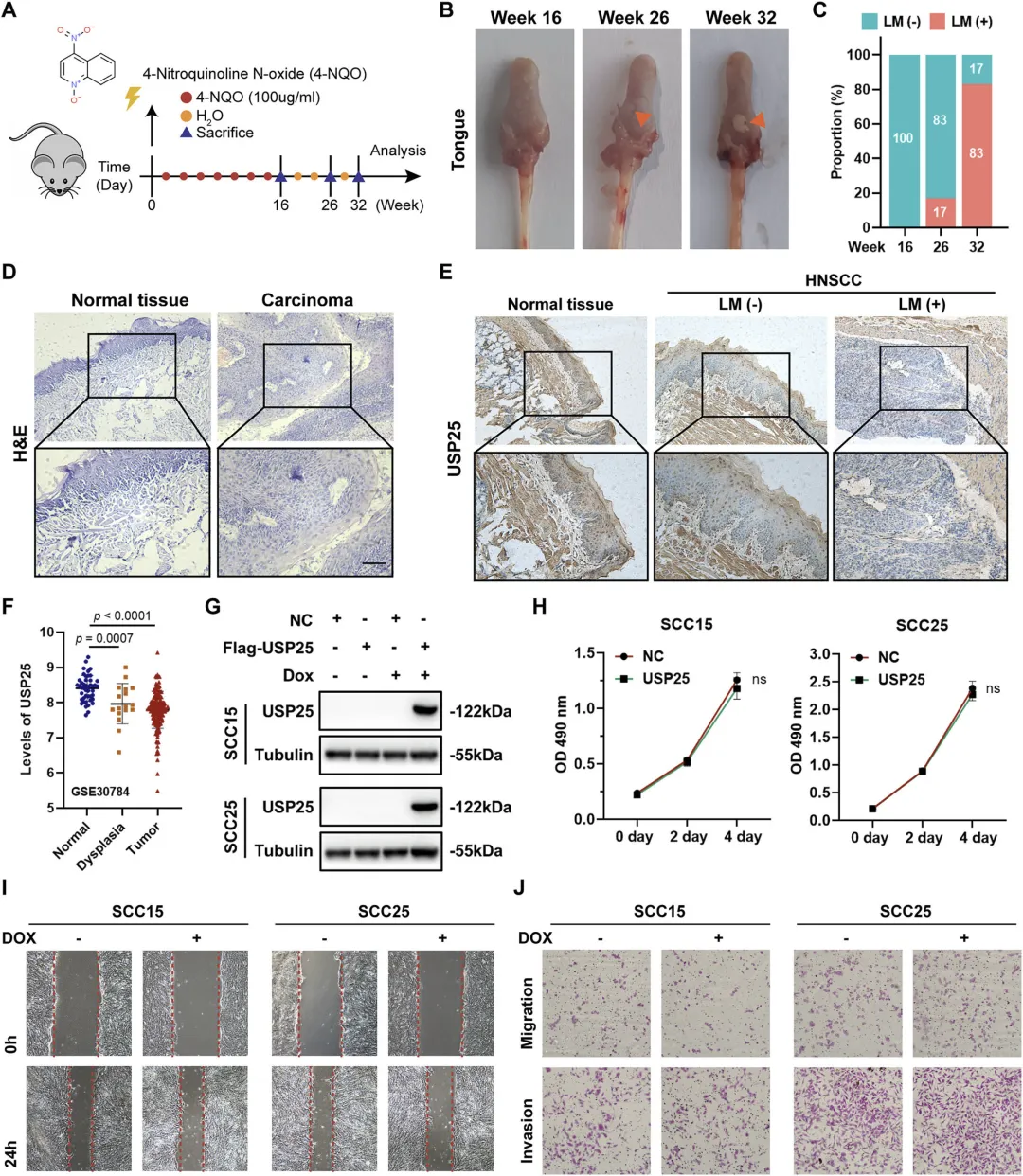
Figure 2:USP25 在体内调控 HNSCC 的恶性进展但不直接影响肿瘤细胞体外生物学功能
该图通过体内动物模型和体外细胞实验,明确了 USP25 对 HNSCC 进展的调控作用及特点。研究者构建 4-NQO 诱导的小鼠 HNSCC 模型,在 16、26、32 周收集舌组织标本,发现随着肿瘤体积增大,淋巴结转移率逐渐升高,HE 染色和 IHC 结果显示,癌组织中 USP25 表达较正常组织显著下调,且发生淋巴结转移的肿瘤组织中 USP25 表达最低;GEO 数据库(GSE30784)分析进一步证实,从正常组织、不典型增生组织到癌组织,USP25 表达逐渐降低,提示其下调是恶性转化的早期且持续事件。体外实验中,在正常 HOK 细胞中敲低 USP25 后,细胞增殖和迁移未受显著影响;在 HNSCC 细胞系(SCC15、SCC25)中通过慢病毒转导稳定过表达 USP25 后,CCK8、克隆形成实验显示细胞增殖无明显差异,伤口愈合和 Transwell 实验也表明细胞迁移和侵袭能力未发生显著改变,说明 USP25 主要在体内调控 HNSCC 的恶性进展,而非直接影响肿瘤细胞的体外生物学功能。
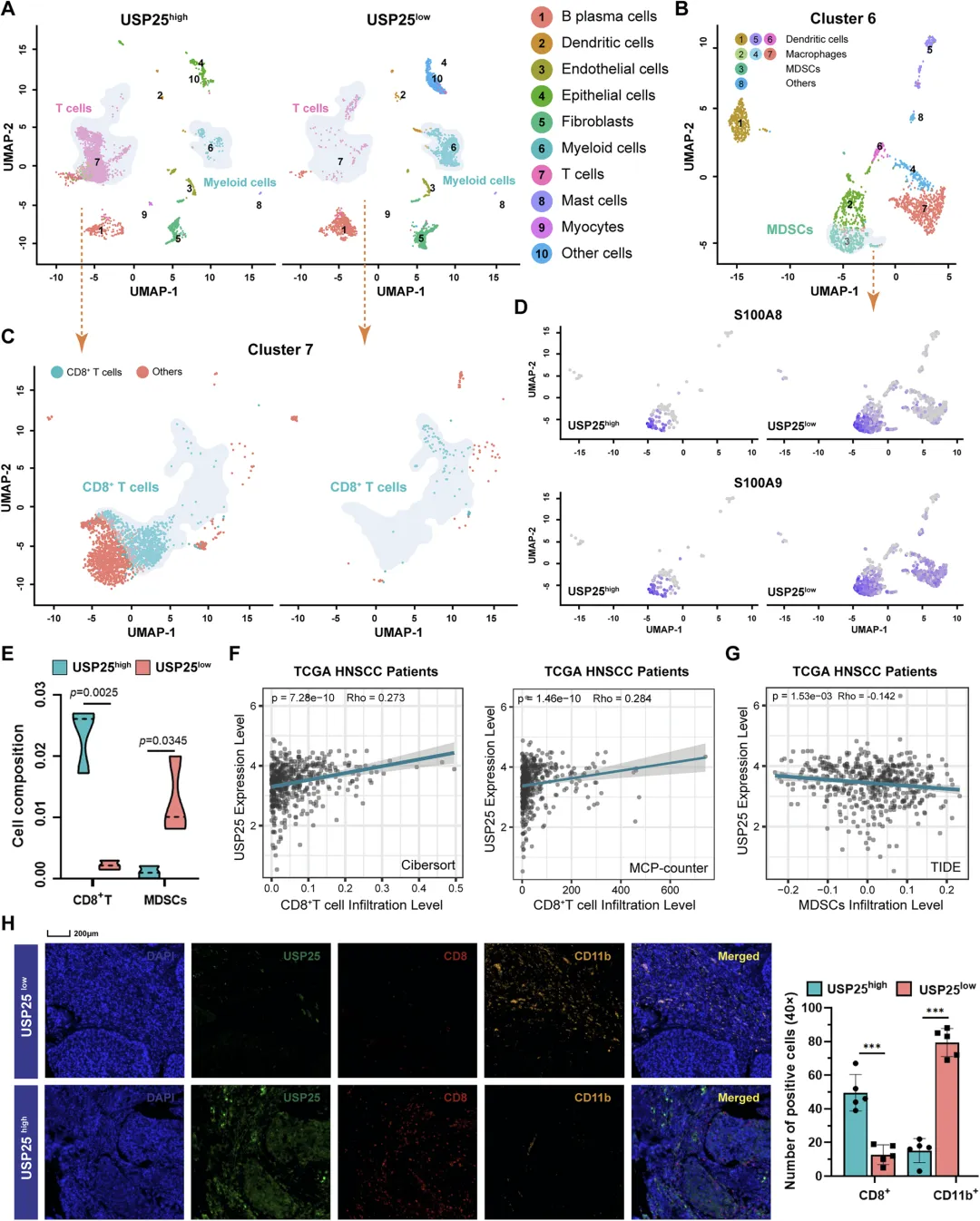
Figure 3:USP25 低表达与 HNSCC 的免疫抑制性肿瘤微环境相关
该图借助单细胞测序和多种验证手段,揭示了 USP25 表达与 HNSCC 肿瘤免疫微环境(TIME)的关联。通过分析 GEO 数据库(GSE181919)的单细胞 RNA 测序数据,经 UMAP 降维和无监督聚类,将髓系细胞分为树突状细胞、巨噬细胞、髓系来源抑制细胞(MDSCs)等 8 个亚群,将 T 细胞分为 CD8+T 细胞及其他亚群,发现 USP25 低表达组的 T 细胞比例显著降低,而 MDSCs 比例升高。MDSCs 的特征标志物 S100A8、S100A9 在 USP25 低表达的肿瘤 MDSC 亚群中富集,提示 USP25 低表达可能通过诱导 MDSCs 积累损害 CD8+T 细胞的抗肿瘤免疫功能。此外,TIMER 数据库分析显示,USP25 表达与 HNSCC 患者肿瘤组织中 CD8+T 细胞浸润呈正相关,与 MDSCs 浸润呈负相关;组织芯片的 mIHC 染色结果进一步验证,USP25 低表达的肿瘤组织中 CD8+T 细胞浸润减少、MDSCs 浸润增加,证实 USP25 在调控 HNSCC 免疫抑制性微环境中发挥关键作用。
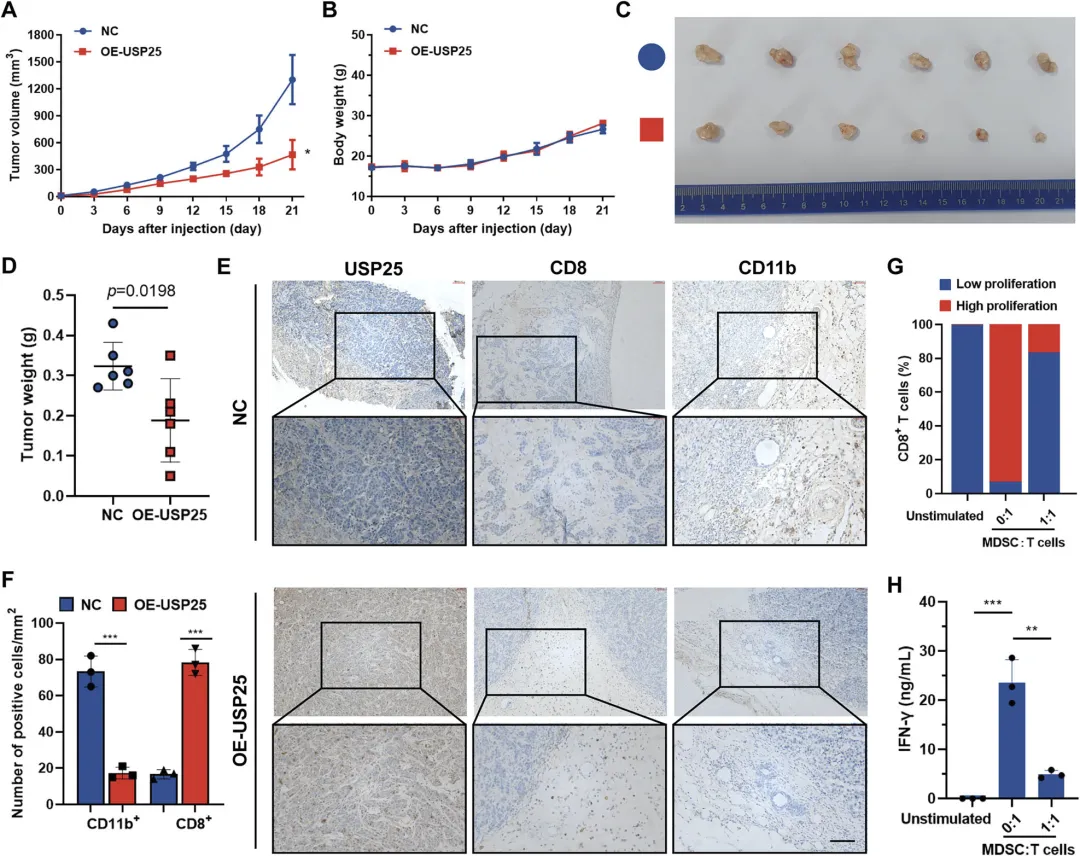
Figure 4:USP25 过表达减少 MDSCs 数量并增加细胞毒性 T 细胞浸润
该图通过小鼠同源肿瘤模型验证了 USP25 对肿瘤免疫微环境的调控效应。研究者构建稳定过表达 USP25 的 HNSCC 同源肿瘤模型,发现与对照组相比,USP25 过表达组的肿瘤体积和重量显著减小,而小鼠体重无明显变化,表明 USP25 过表达可抑制体内肿瘤生长且无明显毒性。IHC 染色检测显示,USP25 过表达的肿瘤组织中,CD8+T 细胞浸润增加,而 MDSCs(CD11b + 标记)浸润减少;功能实验进一步证实,从 USP25 过表达组肿瘤中分离的 MDSCs,对 T 细胞增殖和 IFN-γ 分泌的抑制作用显著弱于对照组,说明 USP25 过表达可逆转肿瘤免疫抑制状态,构建免疫激活型抗肿瘤微环境,其机制与减少 MDSCs 浸润、促进细胞毒性 T 细胞浸润相关。
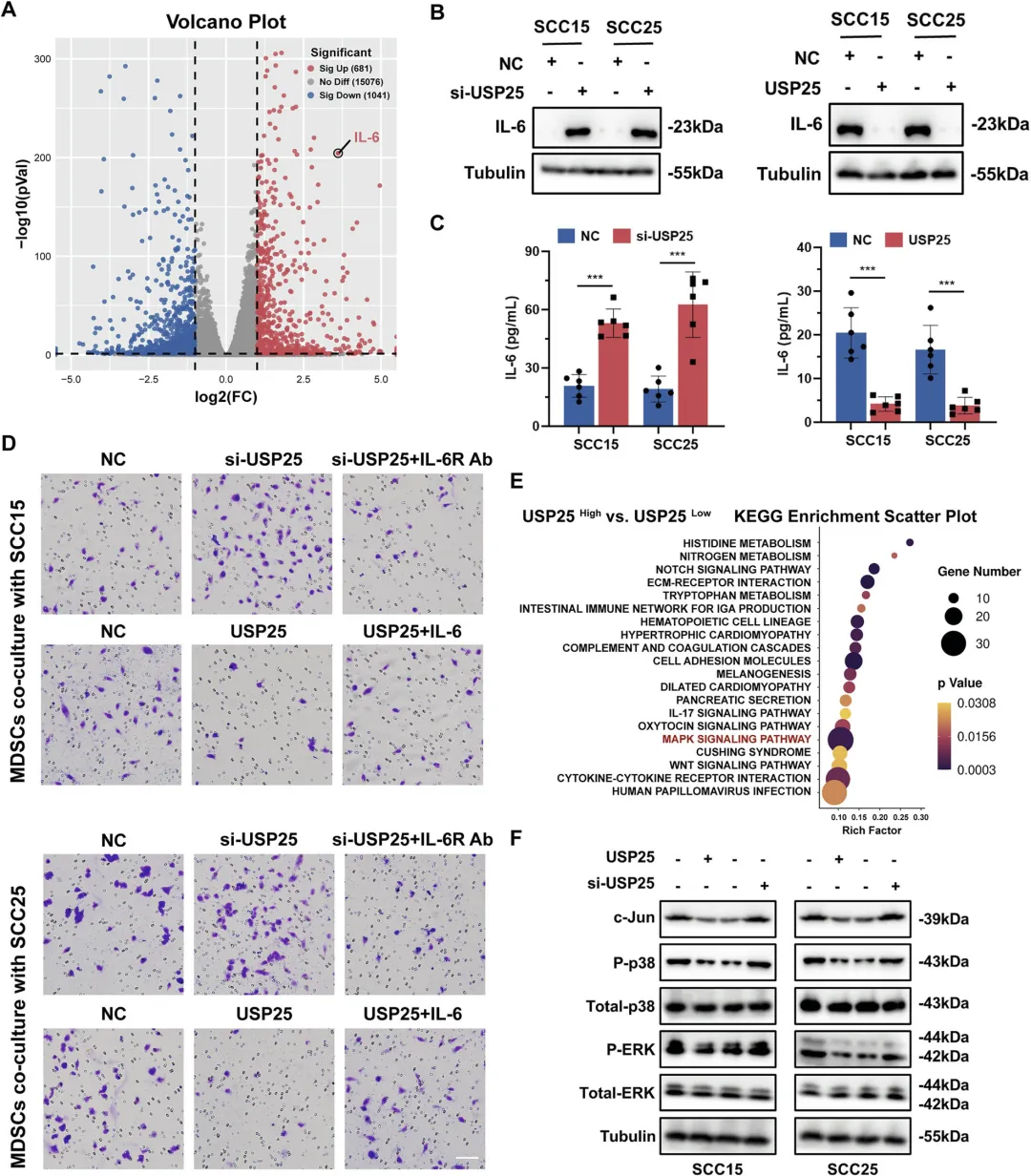
Figure 5:USP25 上调通过抑制 MAPK 通路激活和 IL-6 分泌,减弱 MDSCs 迁移
该图深入探究了 USP25 调控 MDSCs 浸润的分子机制。通过对 USP25 过表达细胞和对照组细胞进行 RNA-seq,筛选出差异表达的可溶性因子,发现 IL-6 是差异最显著的细胞因子;Western blot 和 ELISA 实验证实,USP25 过表达可显著降低 HNSCC 细胞(SCC15、SCC25)中 IL-6 的 mRNA 和蛋白表达及分泌水平,而敲低 USP25 则呈现相反效应。Transwell 趋化实验显示,与 USP25 低表达的肿瘤细胞共培养时,MDSCs 的趋化活性增强,而加入 IL-6 受体抗体可阻断这一效应;反之,与 USP25 过表达的肿瘤细胞共培养时,MDSCs 趋化活性减弱,加入 IL-6 则可逆转该趋势。此外,RNA-seq 的 KEGG 富集分析显示,MAPK 信号通路在 USP25 低表达组显著激活;Western blot 证实,USP25 过表达可抑制 HNSCC 细胞中 MAPK 通路的激活(降低 P-p38、P-ERK 水平)及 IL-6 转录因子 AP-1(c-Jun 亚基)的表达,表明 USP25 通过抑制 MAPK-IL-6 通路,减弱 MDSCs 的迁移能力。
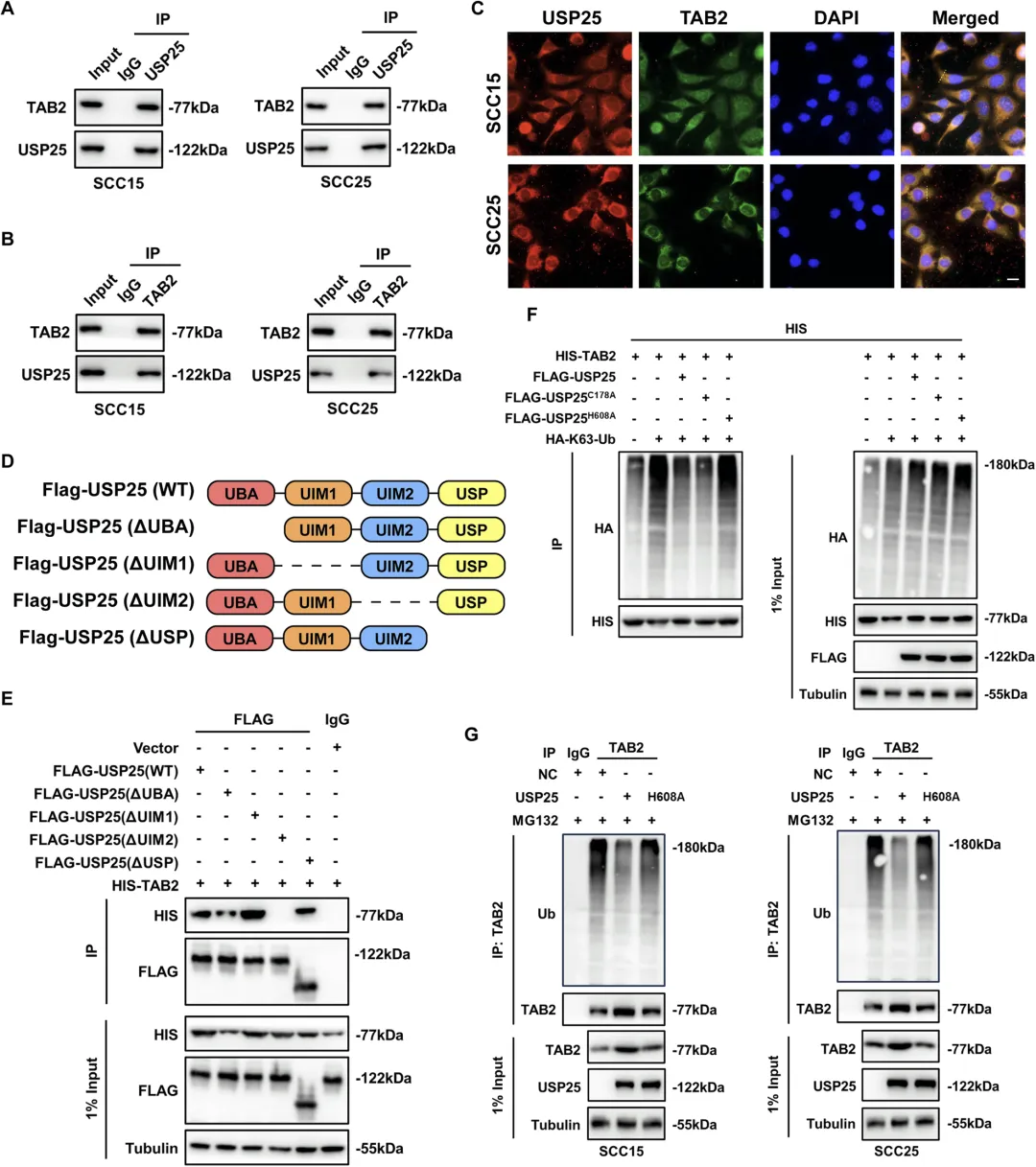
Figure 6:USP25 通过 UIM2 结构域结合 TAB2 并去除其 K63 连接泛素链,抑制 MAPK 信号传导
该图明确了 USP25 调控 MAPK 通路的分子靶点及作用方式。免疫沉淀(Co-IP)和免疫荧光共定位实验证实,USP25 与 TAB2 存在直接相互作用,而不与 TAK1、TAB3 等其他上游信号分子结合。通过构建缺失不同结构域的 USP25 突变体进行 Co-IP 实验,发现 USP25 通过其 UIM2 结构域与 TAB2 发生物理结合。进一步的泛素化实验显示,TAB2 的 K63 连接泛素化对其激活下游 MAPK 信号至关重要,而野生型 USP25 和 C178A 突变体可显著降低 TAB2 的 K63 泛素化水平,H608A 突变体则无此效应,表明 USP25 的去泛素化酶活性依赖于 H608 残基。体外去泛素化实验进一步证实,稳定表达 USP25 的 HNSCC 细胞中,内源性 TAB2 的 K63 泛素化水平显著升高,综合表明 USP25 通过 UIM2 结构域结合 TAB2,去除其 K63 连接泛素链,从而抑制 MAPK 信号通路的激活。
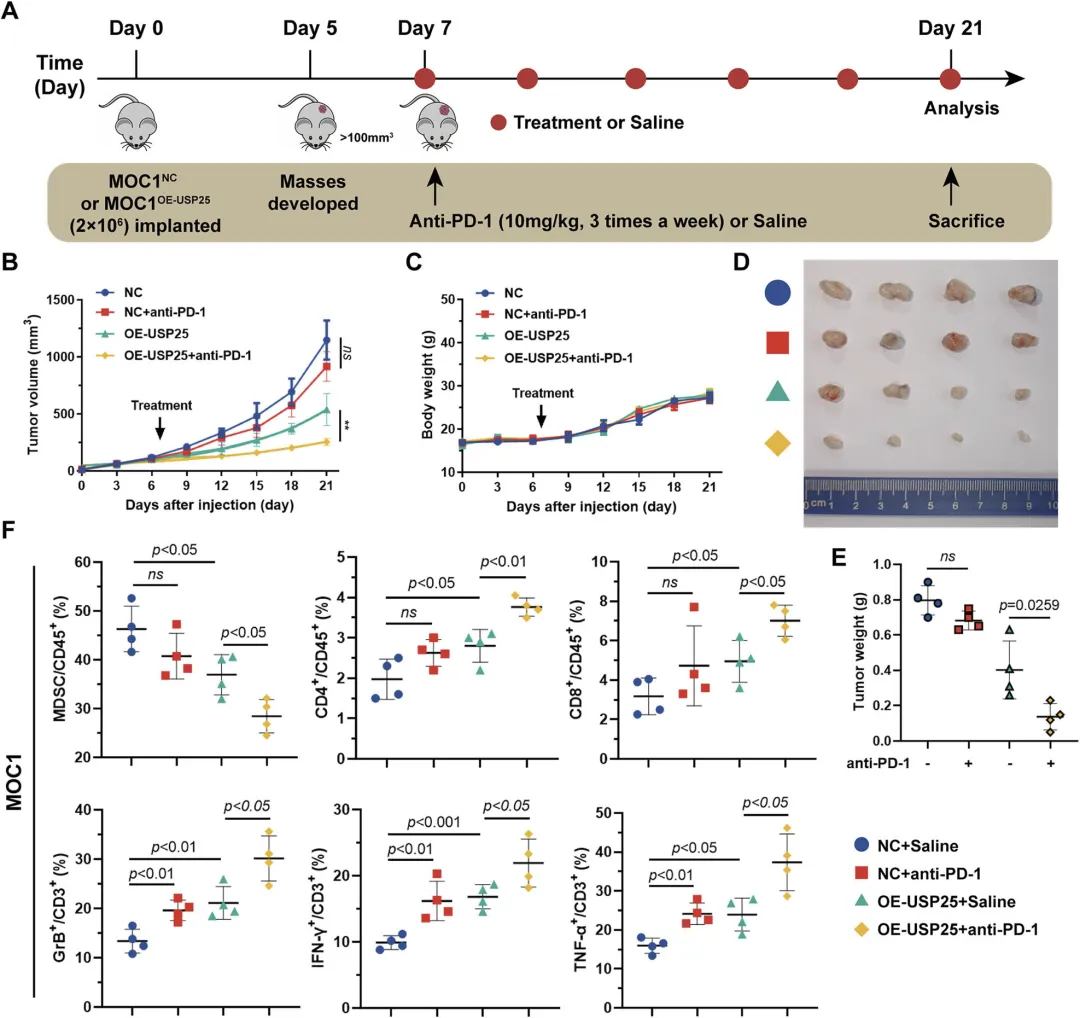
Figure 7:USP25 过表达增强 HNSCC 对 PD-1 抑制剂的体内治疗敏感性
该图验证了 USP25 过表达对 HNSCC 免疫治疗疗效的影响。研究者构建 MOC1 细胞(小鼠口腔鳞癌细胞)的同源肿瘤模型,将 MOC1 NC(对照)和 MOC1 OE-USP25(USP25 过表达)细胞分别植入小鼠体内,待肿瘤体积达到 50-100mm³ 后,给予 PD-1 抑制剂(10mg/kg)或生理盐水治疗。结果显示,USP25 过表达组的肿瘤体积和重量显著小于对照组,且联合 PD-1 抑制剂治疗后,肿瘤抑制效果更显著,而各组小鼠体重无明显差异,表明该联合方案无明显毒性。流式细胞术(FC)分析显示,USP25 过表达联合 PD-1 抑制剂治疗组中,肿瘤组织内 MDSCs 积累减少,而功能性 CD8+T 细胞(包括 Granzyme B+、IFN-γ+、TNF-α+ 亚群)数量显著增加,说明 USP25 过表达可通过改善肿瘤免疫微环境,增强 HNSCC 对 PD-1 抑制剂的治疗敏感性。
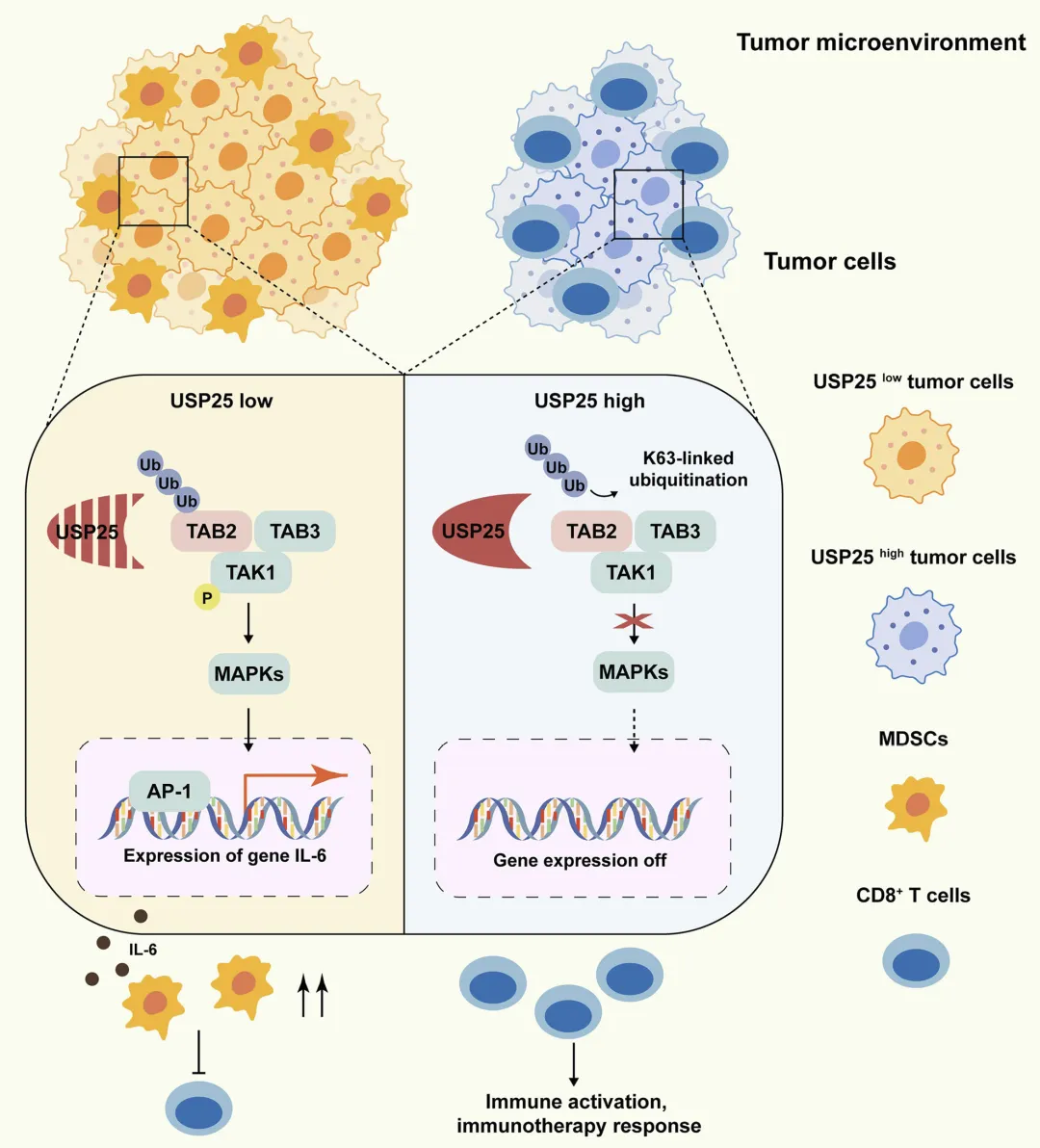
Figure 8:USP25 调控 HNSCC 免疫抑制微环境的分子机制示意图
该图以 schematic 形式清晰呈现了 USP25 在 HNSCC 中的作用机制及调控网络。当肿瘤细胞中 USP25 低表达时,TAB2 的 K63 连接泛素化水平升高,进而激活 TAK1 介导的 MAPK 信号通路,促进转录因子 AP-1 活化,上调 IL-6 的基因表达和分泌;IL-6 作为趋化因子,会招募 MDSCs 进入肿瘤微环境,而 MDSCs 会抑制 CD8+T 细胞的功能,形成免疫抑制性微环境,最终促进肿瘤进展并降低对免疫治疗的应答。当 USP25 高表达时,其通过 UIM2 结构域结合 TAB2,去除其 K63 连接泛素链,抑制 MAPK 通路激活和 IL-6 分泌,减少 MDSCs 迁移和积累,同时促进 CD8+T 细胞浸润并增强其抗肿瘤功能,逆转免疫抑制微环境,从而抑制肿瘤进展并提高对 PD-1 抑制剂的治疗敏感性。
4.结论
本研究明确 USP25 在 HNSCC 中扮演肿瘤免疫微环境调控因子的角色,其低表达与患者不良预后及免疫抑制性微环境密切相关。机制上,USP25 通过结合 TAB2 并去除其 K63 连接泛素链,抑制 MAPK-IL-6 通路,减少 MDSCs 浸润并促进 CD8+T 细胞功能,从而逆转免疫抑制状态。此外,USP25 过表达可显著增强 HNSCC 对 PD-1 抑制剂的治疗应答。综上,USP25 不仅是 HNSCC 预后评估的潜在生物标志物,更可作为免疫治疗联合靶点,为改善晚期 HNSCC 患者免疫治疗效果提供新策略。
局限性:未深入探究 USP25 在不同亚型 HNSCC 中的作用差异,缺乏临床样本中 USP25 与免疫治疗疗效的直接关联数据。展望:未来可扩大临床样本量,验证 USP25 对免疫治疗应答的预测价值;探索 USP25 激动剂与 PD-1 抑制剂的联合治疗方案,为 HNSCC 精准免疫治疗提供新方向。
文章来自:Li X, Jia Y, Zhang R, et al. USP25 attenuates the immunosuppressive tumor microenvironment via the deubiquitination of TAB2 in head and neck squamous cell carcinoma. Cell Death Discov. 2025;12(1):27. Published 2025 Dec 1. doi:10.1038/s41420-025-02883-1
#头颈鳞状细胞癌#USP25#肿瘤免疫微环境#去泛素化#TAB2#MAPK通路#抗PD1治疗#髓系来源抑制细胞
本文中使用的图片来源于Pubmed,因客观原因未能与权利人取得联系。本平台出于学术交流目的引用,无意侵犯原作者权益。如权利人认为不妥,请及时联系公众号后台,我们将立即删除或协商解决。

国家杰青一对一答疑视频
医学省自然申请答疑,立项的关键条件是哪一些?从哪些方向可以杀出重围
临床型博士如何准备国青标书?没有预实验怎么办?专家一对一解答规划
中医药科研研究