第一作者:Chao Lei, Wen Lu, Tingting Shen
通讯作者:Donghui Wei (魏东辉), Yan-Na Ma (马艳娜), Xuenian Chen (陈学年)
通讯单位:郑州大学、河南师范大学
研究要点
本研究开发了一种钯(II)与手性磷酸(CPA)协同催化的新型不对称转化反应,首次实现了碳硼烷与α,β-不饱和羧酸的高效、高选择性“反Michael型”加成(图1)。该反应在温和条件下(0°C,空气氛围),以六氟异丙醇(HFIP)为溶剂,将碳硼烷选择性地引入到不饱和羧酸的α位碳上,成功构建了具有重要生物活性的α-手性羧酸结构单元。反应不仅对邻位、间位和对位碳硼烷表现出优异的B(9)位点选择性,而且对多种官能团化的不饱和羧酸底物展现出广泛的耐受性和出色的对映选择性(ee值高达99%)。结合深入的机理实验与密度泛函理论(DFT)计算,研究团队揭示了反应经由协同金属化-去质子化(CMD)途径实现B-H键活化,并形成关键的五元钯杂环中间体,从而决定了反应的α-区域选择性和立体选择性。这项工作为催化不对称反Michael加成提供了开创性范例,并为合成含碳硼烷的手性羧酸类化合物开辟了新途径。
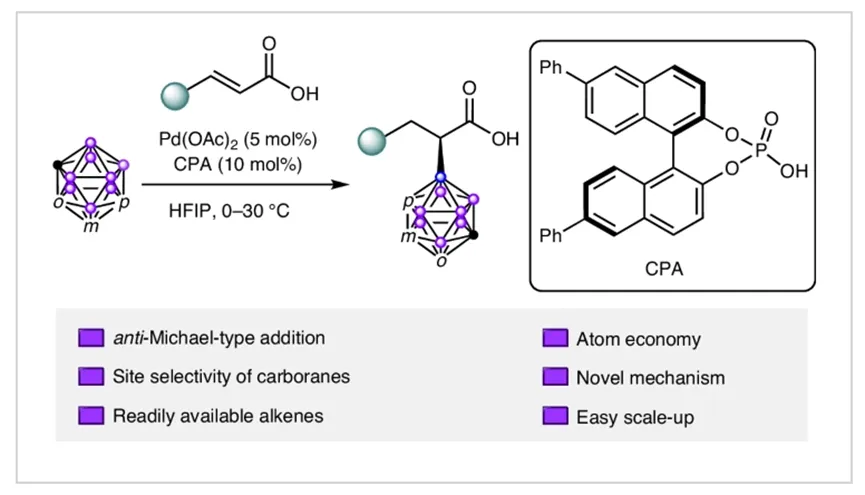 图1. 催化策略示意图
图1. 催化策略示意图
一、研究背景
碳硼烷是一类具有三维芳香性的二十面体硼簇化合物,在材料化学、有机合成和药物研发中展现出巨大潜力。尤其在药物化学领域,碳硼烷衍生物因其能形成独特的二氢键并靶向生物受体的疏水位点而备受关注,同时也是硼中子俘获疗法(BNCT)的候选材料。因此,发展将碳硼烷高效、高选择性地引入有机分子的新方法具有重要价值。
尽管碳硼烷笼碳原子的官能化已取得长足进展,但笼硼原子(B-H键)的精准修饰,特别是B-H键的不对称转化,仍然是一个艰巨挑战。这主要源于B-H键极性弱,且在邻位和间位碳硼烷中存在多达十个化学环境相似的B-H键,导致选择性控制困难。更重要的是,碳硼烷的硼取代基团通常表现为富电子性质,这与碳取代基的吸电子特性截然不同,使得相应衍生物具备独特的性质。因此,发展选择性B-H官能化,尤其是催化不对称方法,将手性碳中心直接与硼顶点相连,对于拓展碳硼烷在不对称合成和手性药物中的应用具有里程碑意义。
在传统的有机合成中,Michael加成是构建β-手性中心最有力的工具之一。相比之下,其逆向过程——即亲核试剂进攻α,β-不饱和羰基化合物的α位(“反Michael”或“α-加成”)——则鲜有报道。鉴于α-手性羰基化合物在生物活性分子和药物中的核心地位,发展催化不对称的反Michael加成反应极具吸引力(图2)。
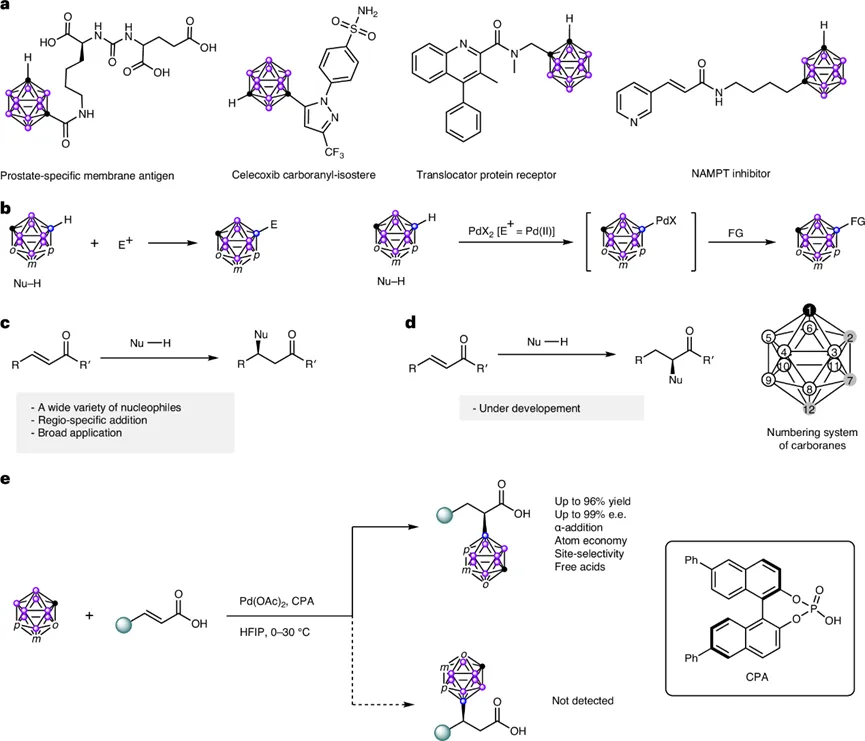 图2. 硼烷及其在催化不对称Michael反应与反Michael加成反应中的应用
图2. 硼烷及其在催化不对称Michael反应与反Michael加成反应中的应用
二、研究思路
1. 催化剂设计与反应条件优化
基于前期对碳硼烷亲电取代反应的研究(特别是在HFIP溶剂中,钯(II)可作为软亲电试剂高效活化碳硼烷的B-H键),研究团队设想:能否在钯(II)和手性配体催化下,利用碳硼烷作为独特的“Michael供体”,与α,β-不饱和羰基化合物发生不对称加成?
为此,他们选择反应性适中且在B(9)位官能化中表现出良好选择性的间位碳硼烷(m-carborane)作为模型底物,与4-硝基肉桂酸进行反应探索。在Pd(OAc)₂存在下,系统筛选了一系列布朗斯特酸作为手性配体(图3)。研究发现,先前报道用于促进Pd(II)催化不对称C-H活化的手性氨基酸只能得到外消旋产物。而手性联萘二羧酸(L2)和联萘酚磷酸(L3)则能带来一定对映选择性。随后,一系列手性磷酸(CPA)衍生物(L4-L15)被考察。结果表明,在BINOL骨架6,6‘位引入芳基取代基对获得立体控制至关重要。最终,配体6,6’-二苯基联萘酚磷酸(L11)脱颖而出,在优化的反应条件下(Pd(OAc)₂ 5 mol%, L11 10 mol%, HFIP溶剂,0°C),能以87%的收率和95%的ee值得到目标产物1。值得注意的是,具有强配位能力的双膦配体(如BINAP)未能得到任何产物,表明需要一个缺电子的Pd(II)催化中心来启动反应。
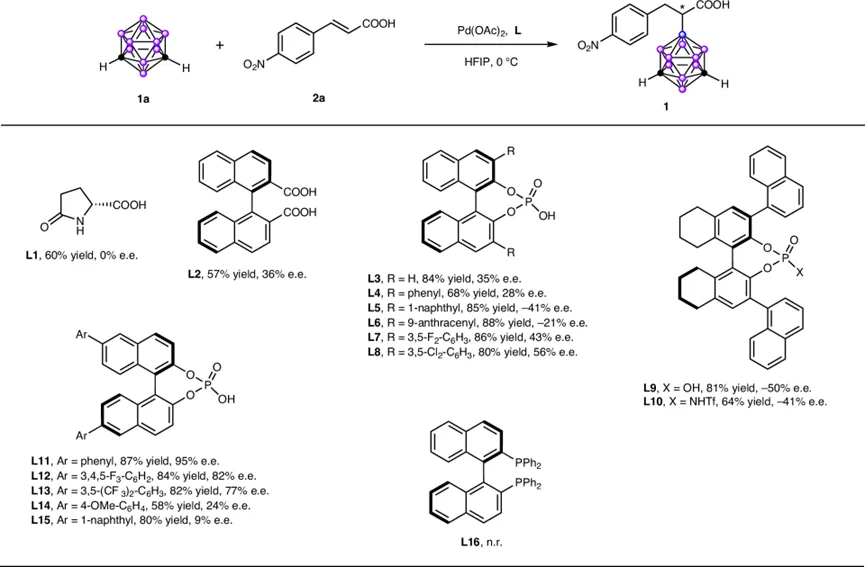 图3. 配体的筛选
图3. 配体的筛选
2. 底物普适性考察:广泛的官能团耐受性
在确立最优条件后,研究团队系统考察了α,β-不饱和羧酸的底物范围(图4)。各种对位、间位和邻位取代的肉桂酸衍生物均能顺利反应,以中等至优秀的收率(42%-92%)和71%-95%的ee值得到产物1-25。有趣的是,芳环上带有吸电子基团的底物通常表现更优;而强供电子基团(如15)则抑制了反应。特别值得关注的是,对甲酰基和硼酸酯基等敏感官能团在该反应条件下能完好保留。
对于氮杂芳环取代的不饱和羧酸,标准条件下因氮原子与钯的强配位作用而无法反应。通过加入三氟甲磺酸(HOTf)保护氮原子,成功获得了产物26和27。β-烷基取代的丙烯酸也是合适的底物,长烷基链有利于提高对映选择性(28-30)。王浆酸及其衍生物也能顺利转化(31, 32)。此外,β位带有羰基、氯等吸电子基的底物(34-36)仍能获得α-加成产物,证明了反应独特的区域选择性。该反应还展示了出色的后期官能化潜力,一系列生物活性分子和天然产物(如吲哚、酪氨酸、薄荷醇、半乳糖等)经衍生化为肉桂酸后,均能高效转化为目标产物38-45,ee值在62%-93%之间。
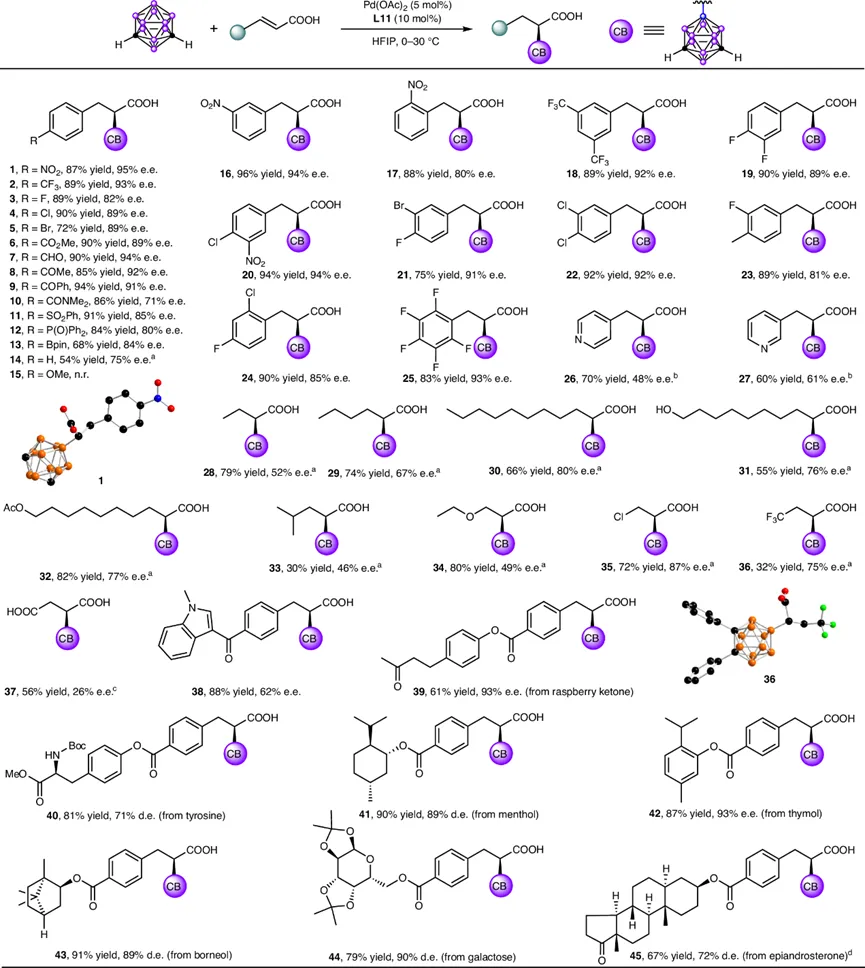 图4. α,β-不饱和羧酸的底物范围
图4. α,β-不饱和羧酸的底物范围
3. 碳硼烷底物的广泛适用性
研究进一步拓展至不同结构和取代模式的碳硼烷(图5)。与间位碳硼烷类似,邻位碳硼烷(o-carborane)也能以77%的收率和96%的ee值,以及优异的B(9)选择性(B9/B8比例约9:1)得到产物46。一系列双碳取代的邻位碳硼烷(47-65)均能顺利反应,收率37%-91%,ee值高达91%-99%。值得注意的是,硅基取代的底物(51)在反应中硅基得以保留,这与通常观察到的Pd(II)催化B-H活化中硅基消除的现象形成鲜明对比。单碳取代的邻位碳硼烷则生成B(8), B(9), B(10), B(12)的混合物,其中主产物B(9)加成物(66, 67)仍能以高对映选择性分离。硼顶点上的取代基对反应的收率和对映选择性均有显著影响(68-74)。此外,该策略甚至适用于反应性更低的、通常被视为惰性的对位碳硼烷(p-carborane),以55%收率和78% ee值得到产物78。
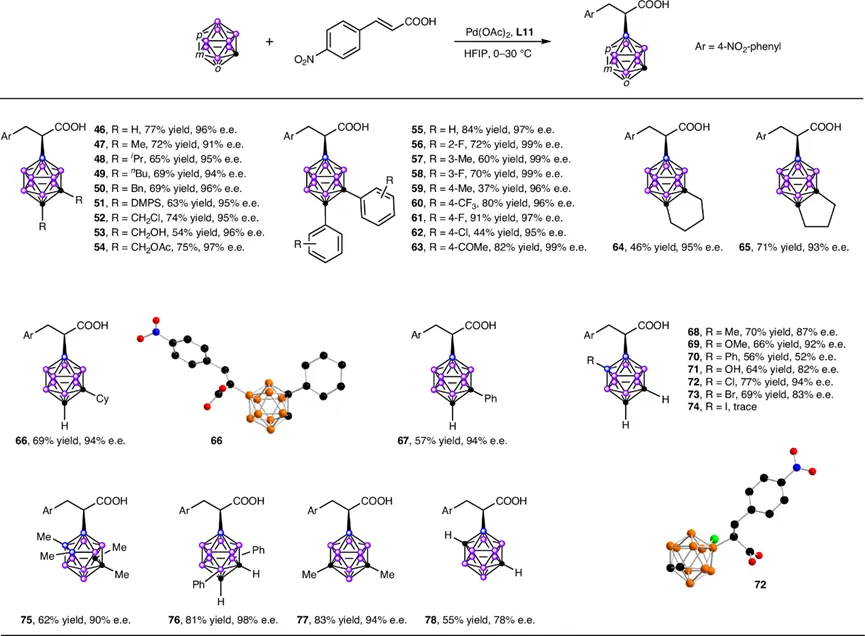 图5. 硼烷的底物范围
图5. 硼烷的底物范围
4. 合成应用展示
该反应的实用性和可放大性得到了验证(图6)。邻位碳硼烷与4-硝基肉桂酸的克级规模反应,能以86%的收率和93%的ee值获得产物46。使用¹⁰B富集的碳硼烷为底物,能以90%收率和95% ee获得相应的¹⁰B标记产物,展示了其在BNCT相关药物合成中的潜力。
此外,所得手性α-碳硼烷基羧酸产物中的羧基和硝基可进行多样化衍生,得到相应的醇(79)、酯(80)、酰胺(81)和胺(82)等,且在这些转化过程中手性中心的对映体纯度得以保持,证明了该手性中心的化学稳定性。
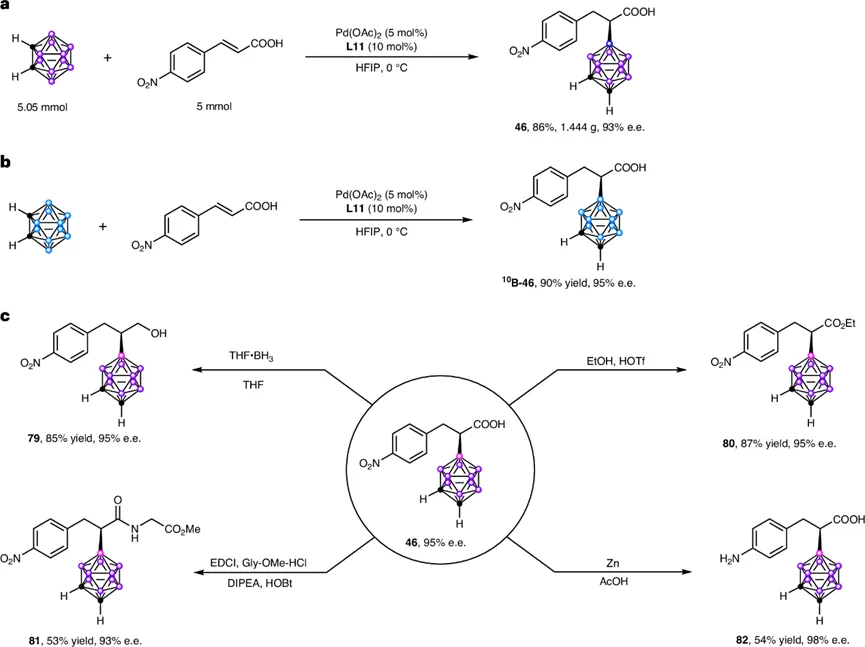 图6. 合成应用
图6. 合成应用
5. 反应机理的深度剖析
控制实验与氘代标记研究
为探究反应机理,研究人员进行了一系列控制实验(图7a)。将(E)-4-硝基肉桂酸换为(Z)-异构体,对映选择性急剧下降,表明(E)构型对获得高手性控制至关重要。使用肉桂酸酯或肉桂酰胺替代游离羧酸时,反应虽能进行,但ee值显著降低(33%和17%)。若无手性配体,仅得到外消旋产物。在肉桂酸酯或肉桂酰胺的体系中额外加入乙酸,能恢复部分产率。这些结果共同表明,羧基质子的存在是实现高效、高选择性转化的关键。
氘代标记实验揭示了β位氢原子的来源(图7b)。向体系中加入DOAc或D₂O,氘原子并未掺入产物。然而,当使用氘代碳硼烷(B(8,9,10,12)-D₄-o-carborane)进行反应时,氘原子通过顺式加成过程专一地出现在产物的β位,且硼顶点上也发生了H/D交换。使用全氘代硼顶点的碳硼烷进行反应,进一步证实了β位氘原子来源于碳硼烷骨架。动力学同位素效应(KIE)值为3.5,表明B-H键的断裂是反应的决速步骤。
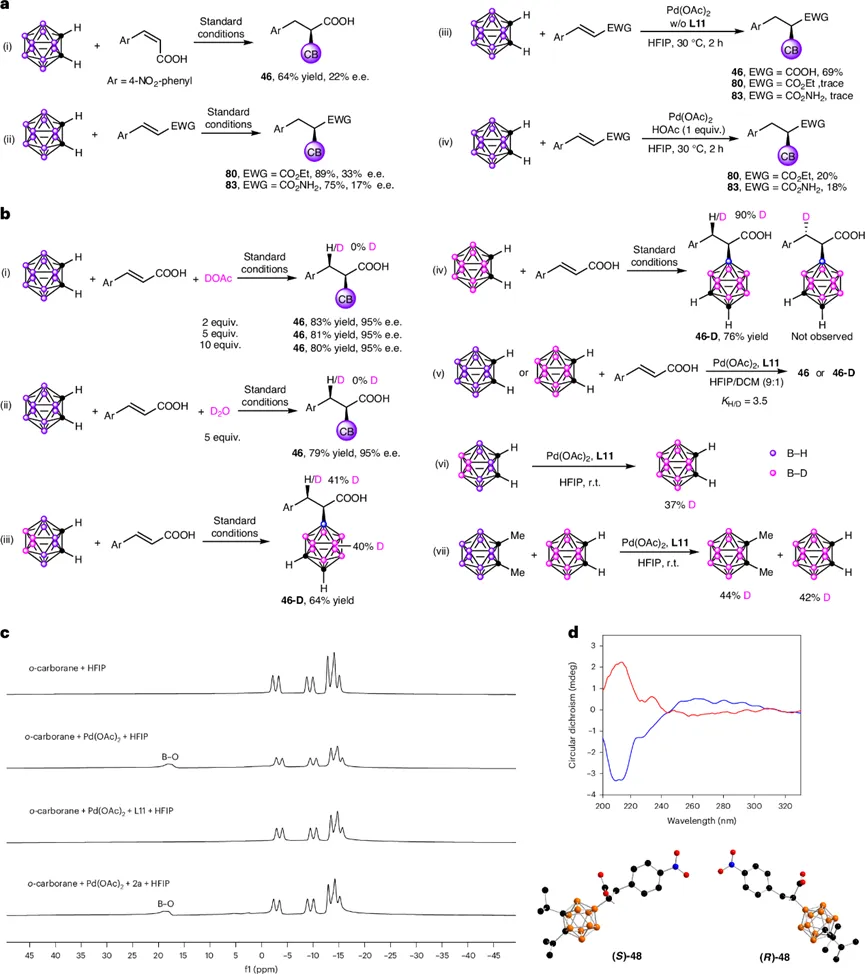 图7. 机制探究
图7. 机制探究
计算化学揭示选择性起源
通过DFT计算(图8, 9),研究团队比较了生成α和β加成产物的三种可能路径:协同金属化-去质子化(CMD)路径、协同氢转移与Pd-B键形成路径以及生成Pd-H物种的B-H插入路径。计算表明,CMD路径在能量上最为有利。
在CMD路径中,Pd(OAc)L11首先通过CMD过渡态(TS1)活化碳硼烷的B-H键,形成中间体M1。随后,关键的C-B键形成步骤决定了区域和对映选择性。计算发现,生成α-加成产物的过渡态(TS2S-α和TS2R-α)在能量上显著低于生成β-加成产物的过渡态(TS2S-β和TS2R-β),这与实验观察到的α-区域专一性一致。更重要的是,对于α-加成路径,(S)-构型过渡态TS2S-α的能量比(R)-构型过渡态TS2R-α低约2.4 kcal/mol,这完美解释了反应的高对映选择性。
对关键过渡态TS2S-α和TS2R-α的非共价相互作用分析揭示,前者存在更多更强的弱相互作用(如B-H…π、孤对电子…π相互作用),这些相互作用稳定了TS2S-α,使其在能量上更具优势,从而决定了产物的绝对构型。计算还支持了后续的协同去金属化-质子化步骤,以及形成关键五元钯杂环中间体M2是反应实现α-选择性的关键。
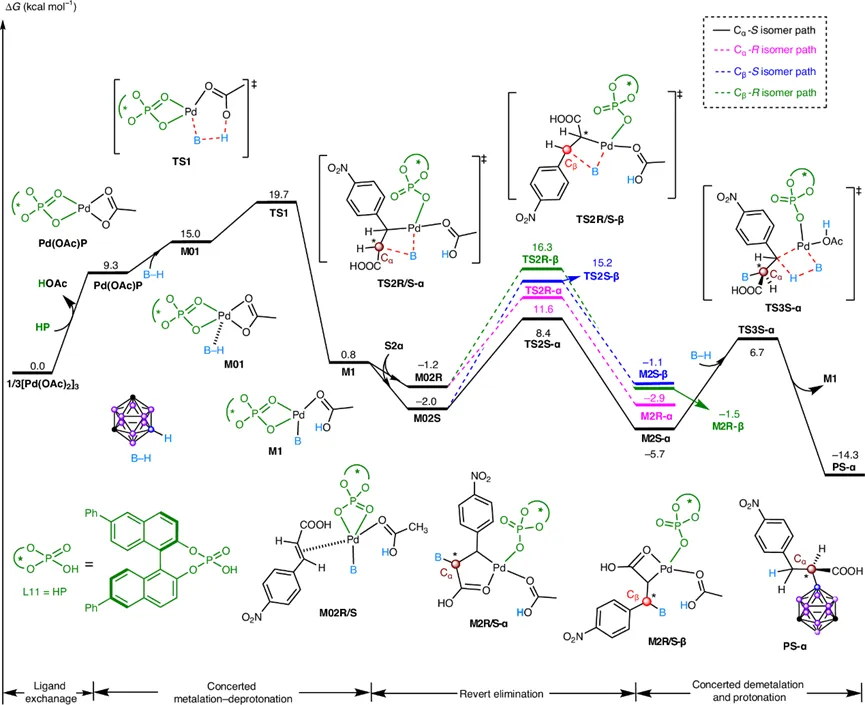 图8. DFT计算
图8. DFT计算
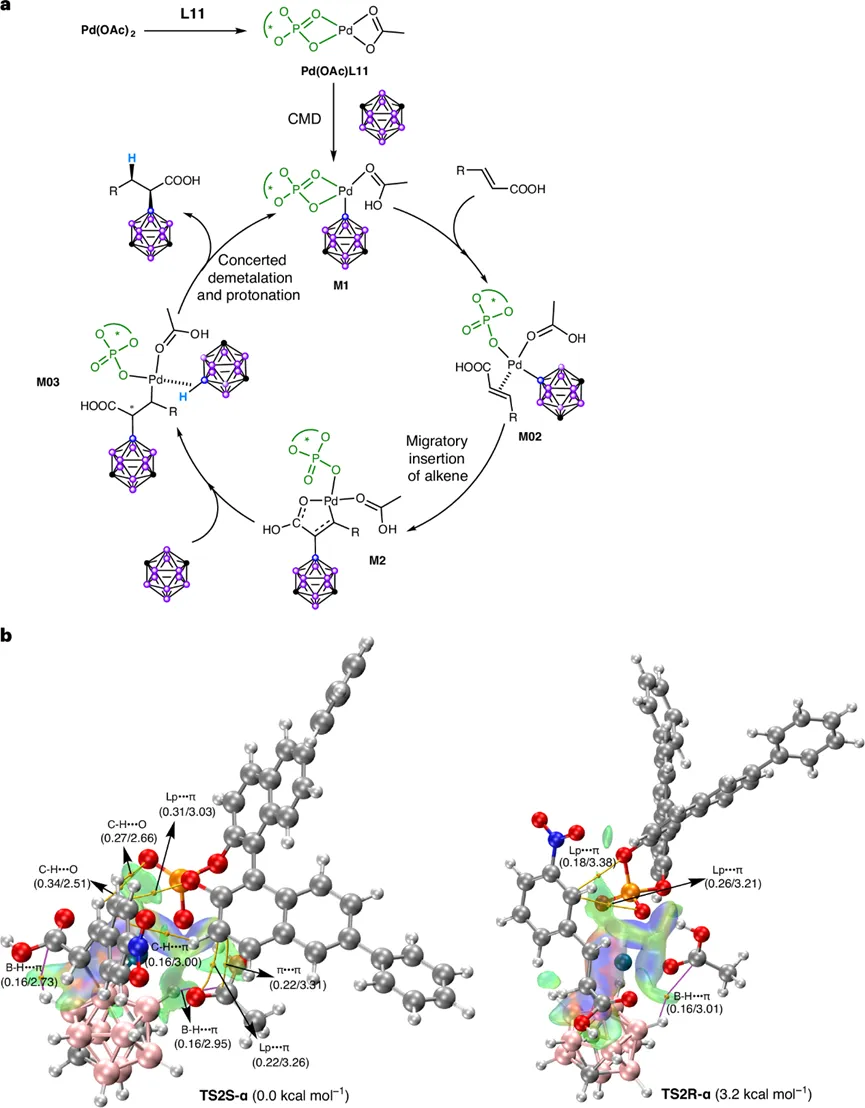 图9. 机制模型
图9. 机制模型
三、小结
本研究成功发展了一种钯(II)/手性磷酸协同催化的碳硼烷与α,β-不饱和羧酸的不对称反Michael加成反应。该反应条件温和,具有优异的B(9)位点选择性、α-区域选择性和对映选择性,底物适用范围广泛。其核心创新在于利用CMD机制实现碳硼烷B-H键的不对称活化,并通过形成关键的五元钯杂环中间体,成功将碳硼烷引入到不饱和羧酸的α位,构建了极具价值的α-手性羧酸骨架。
这项工作不仅为催化不对称反Michael加成反应提供了重要范例,更重要的是,它首次实现了将三维芳香性的碳硼烷簇与经典有机手性中心的精准、不对称连接,为碳硼烷化学和不对称合成领域的交叉融合开辟了新方向。结合详尽的实验机理研究和理论计算,该研究从原子和分子层面阐明了反应选择性起源,为未来理性设计基于碳硼烷的新型手性催化剂和功能分子奠定了坚实的科学基础。未来研究方向可包括拓展亲核试剂类型、探索其他不饱和化合物以及开发更高效的手性催化体系,进一步推动碳硼烷在手性材料、药物化学等领域的应用。
原文详情
Lei, C., Lu, W., Shen, T. et al. Palladium-catalysed asymmetric anti-Michael-type addition of α,β-unsaturated carboxylic acids with carboranes. Nat. Catal.(2026).
https://doi.org/10.1038/s41929-026-01480-4