郑州轻工业大学张治红/杜淼&新乡医学院刘嘉梦,最新AFM!促进硝酸盐合成过程*NOH中间体生成动力学:光辅助电催化氮氧化机制解析研究!
电催化氮氧化反应(eNOR)为硝酸盐(NO₃⁻)合成提供了一条可持续路径,但其实际应用受限于缓慢的中间体生成动力学。虽然水氧化过程中产生的·OH可促进关键中间体·NOH的形成,但·OH与催化位点间的协同机制仍未充分阐明。
2026年02月07日,郑州轻工业大学张治红、杜淼团队在Advanced Functional Materials期刊发表题为“Boosting *NOH Intermediate Generation Kinetics for Nitrate Synthesis: Insights From Photo-Assisted Electrocatalytic Nitrogen Oxidation”的研究论文,团队成员陶正、张帅为论文共同第一作者,张治红、新乡医学院刘嘉梦、杜淼为论文共同通讯作者。
第一作者:陶正、张帅
通讯作者:张治红、刘嘉梦、杜淼
通讯单位:郑州轻工业大学、新乡医学院
论文DOI:10.1002/adfm.202531671
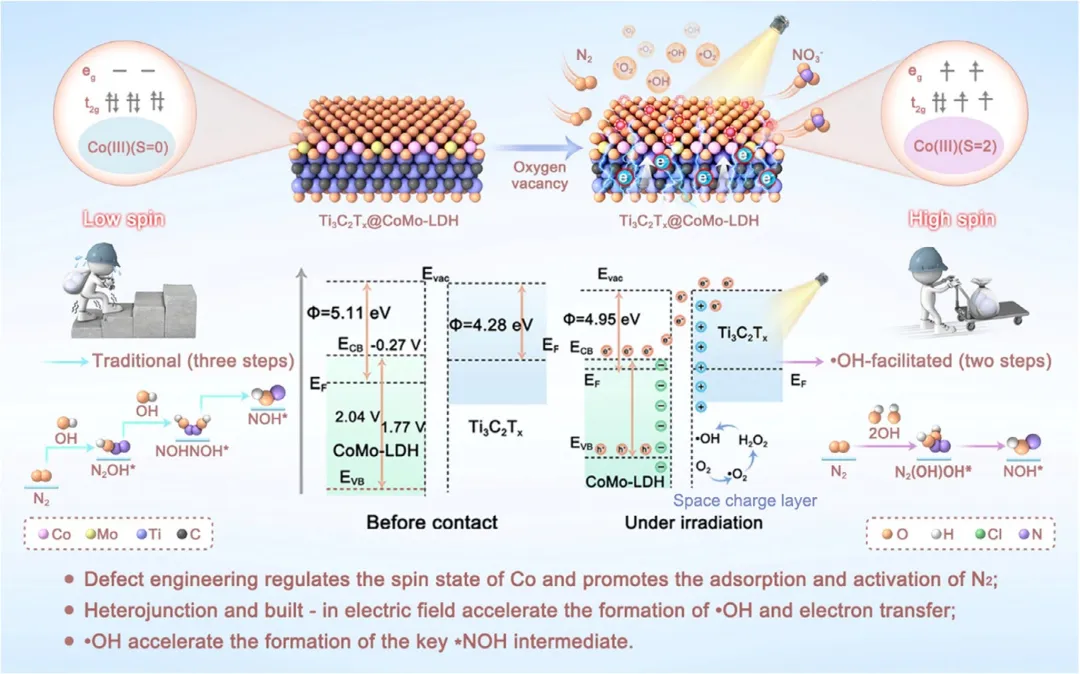
该研究提出一种自旋态调控策略,以增强模拟太阳光照下*NOH的生成,从而显著提升eNOR的NO₃⁻产率与法拉第效率(FE)。研究人员设计了一种异质结催化剂,由在Ti₃C₂Tₓ MXene上原位生长的CoMo基层状双氢氧化物纳米片(CoMo-LDH@Ti₃C₂Tₓ)构成,用于实现光辅助eNOR。Mo掺入Co-LDH与异质结内建电场共同增强了Mo-Co轨道杂化,诱导Co³⁺中心自旋态由t₂g⁶eg⁰构型向t₂g⁴e g²构型转变。这种电子调控强化了N₂活化,并在光照下通过两个·OH自由基的协同参与加速了*NOH形成,从而显著提升了eNOR动力学。因此,CoMo-LDH@Ti₃C₂Tₓ在光照下实现了198.55 µg h⁻¹ mgcat⁻¹的NO₃⁻产率和46.22%的FE,优于暗态条件及现有先进催化剂。这些发现强调了自旋态工程与自由基协同作用在推进可持续硝酸盐生产中的关键作用。
氮(N₂)作为地球大气中最丰富的气体组分(约占78 vol%),是维持生命活动与工业生产的核心元素。然而,其超稳定N≡N(键能941 kJ mol⁻¹)严重限制了其直接转化为生物可利用或工业有用的含氮化合物,构成了全球氮循环中的关键固氮瓶颈。在各种含氮产物中,硝酸(NO₃⁻)及其衍生物硝酸盐在农业、能源储存与精细化工合成中不可或缺,使其可持续生产成为应对全球粮食安全与能源转型挑战的战略重点。传统工业HNO₃合成主要依赖通过氨氧化耦合哈伯-博世法的Ostwald工艺,该工艺需在高能耗条件下进行(400–500°C, 20–30 MPa),消耗大量化石能源,约占全球能源消耗的2%,并产生大量温室气体与空气污染物。当前研究重点关注四种直接实现N₂到NO₃⁻转化的催化策略:光催化、电催化、等离子体处理与微气泡系统。其中,电催化策略在成本效益与可持续性方面表现突出,使其通过氮氧化反应(NOR)合成HNO₃具有前景。电催化NOR(eNOR)在常温常压下运行,以可再生电力为能源输入,直接将N₂转化为NO₃⁻/HNO₃,符合碳中和与绿色制造目标。深入的机理研究表明,表面吸附的羟胺(*NOH)中间体的生成是电催化NOR的速率决定步骤(RDS)。不同于*N₂、*N₂OH等其他中间体,*NOH的形成涉及N₂活化与表面羟基物种(*OH)的耦合,且需克服较高的动力学能垒。当前优化*NOH中间体生成的方法主要集中在调控催化剂电子结构或修饰电解质环境,但进展有限。例如,过渡金属氧化物(如Co₃O₄、NiO)表现出适中的*OH吸附能力,但无法有效活化N₂。MXene基材料显示出优异的导电性,但缺乏足够的*NOH耦合活性位点。这些挑战表明,单一的电子结构调控或活性位点工程方法不足以突破*NOH生成的RDS瓶颈。为应对电催化的动力学限制,光辅助eNOR被提出作为一种协同策略。
在光照下,光生电子/空穴可调控催化剂表面电荷状态,而光激发的活性氧物种(ROS,如·OH、·O₂⁻与¹O₂)可作为电子穿梭体促进中间体转化。近期研究表明,光辅助NOR系统在NO₃⁻生产上表现出比暗态更高的法拉第效率(FE),但光如何调控*NOH生成的内在机制仍不明确。阐明这一机制对推动光辅助电催化在eNOR中的应用具有重要意义。在各种过渡金属电催化剂中,含高自旋态Co位点的钴基材料因其高能未配对电子而备受关注。3d轨道电子排布可增强与N₂分子中π*反键轨道的电子相互作用,从而促进N≡N键断裂。钴基电催化剂表面的结构重构在电催化过程中频繁发生。含高价Co³⁺物种的CoOOH的原位形成通过促进·OH中间体生成来增强水氧化,进而通过eNOR路径促进NO₃⁻电合成。通过增强关键*NOH中间体的生成,开发钴基电催化剂以提高光照下NO₃⁻合成效率具有前景。
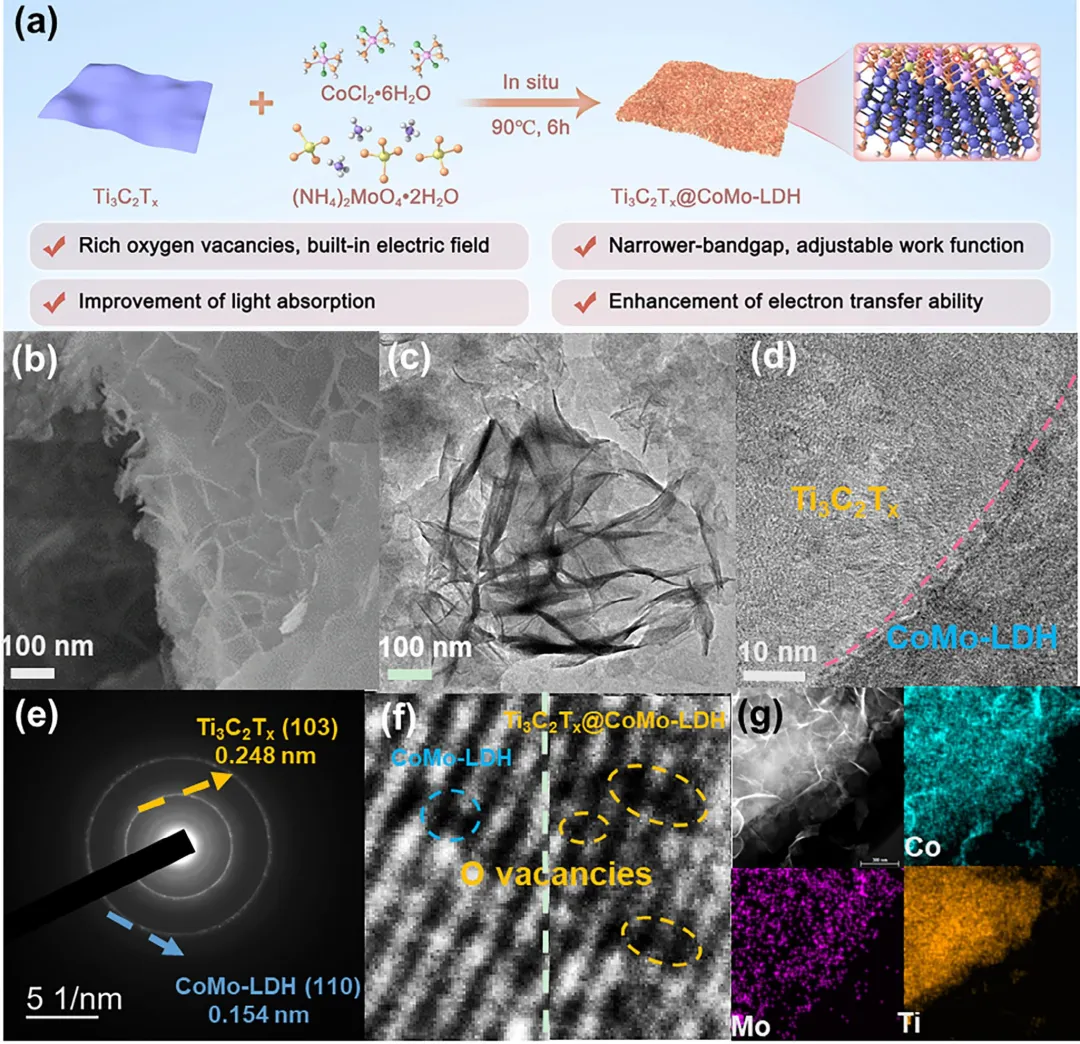
在此,该研究构建了一种Ti₃C₂Tₓ@CoMo-LDH n-n异质结催化剂,用于通过光辅助eNOR进行NO₃⁻电合成,以增强*NOH生成动力学。如图1所示,CoMo-LDH纳米片(NSs)原位生长在Ti₃C₂Tₓ MXene纳米片周围形成n-n异质结。该异质结集成了CoMo-LDH的特征(如氧空位(Oᵥs)、超薄层与活性位点)与Ti₃C₂Tₓ MXene的特性(如优异的光吸收与光催化活性),通过带隙窄化与加速光生载流子转移协同提升ROS生成效率。Ti₃C₂Tₓ@CoMo-LDH异质结在模拟太阳光照射下,于N₂饱和的0.1 M Na₂SO₄中、1.8 V vs. RHE条件下,实现了198.55 µg h⁻¹ mgcat⁻¹的NO₃⁻产率与46.22%的高FE,其产率与FE分别较暗态条件提升了约2.1倍与1.8倍。此外,结合原位表征(如原位拉曼、衰减全反射傅里叶变换红外光谱与微分电化学质谱)与密度泛函理论DFT计算,直接捕获了*NOH的动态演化并量化了其在光电协同下的动力学能垒。该研究不仅阐明了光生·OH与异质结内建电场在促进*NOH形成中的协同机制,还揭示了Co³⁺自旋态转变(低自旋至高自旋)在优化活性位点与N₂/*OH间轨道相互作用中的作用,最终建立了*NOH生成速率与NO₃⁻合成效率间的定量关系,为进一步设计高动力学NOR催化剂提供了理论基础。
图1 eNOR中·OH的重要性示意图及Ti₃C₂Tₓ@CoMo-LDH基异质结通过光辅助电催化增强eNOR的·OH生成策略。
图2 (a) Ti₃C₂Tₓ@CoMo-LDH异质结制备流程示意图。(b) Ti₃C₂Tₓ@CoMo-LDH的SEM图像。(c,d) Ti₃C₂Tₓ@CoMo-LDH的HR-TEM图像。(e) Ti₃C₂Tₓ@CoMo-LDH的SADE图像。(f) CoMo-LDH与Ti₃C₂Tₓ@CoMo-LDH的HR-TEM图像。(g) Ti₃C₂Tₓ@CoMo-LDH的EDS mapping图像(Co:青色,Mo:紫色,Ti:橙色)。
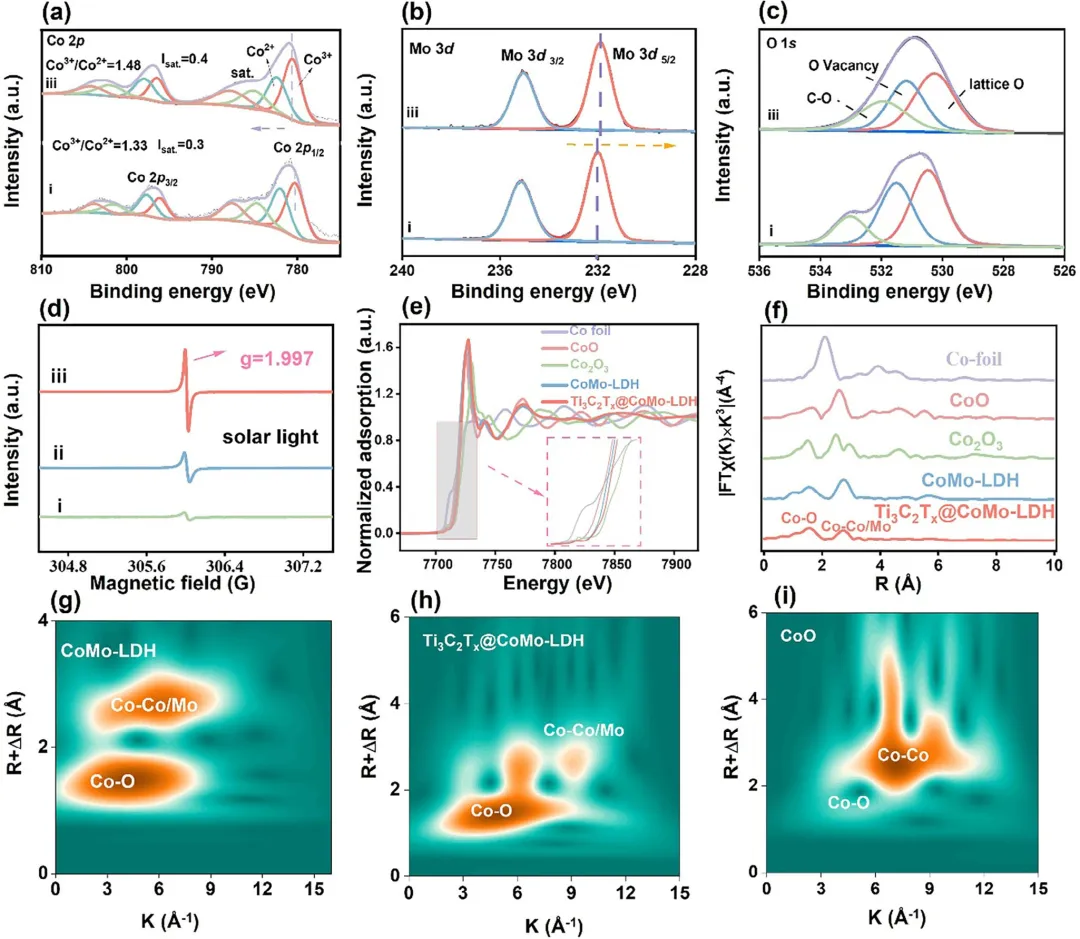
图3 (i) CoMo-LDH、(ii) Ti₃C₂Tₓ与(iii) Ti₃C₂Tₓ@CoMo-LDH的高分辨率(a) Co 2p、(b) Mo 3d和(c) O 1s XPS谱图。(d) (i) CoMo-LDH、(ii) Ti₃C₂Tₓ与(iii) Ti₃C₂Tₓ@CoMo-LDH在太阳光辅助下的EPR谱图。(e) Ti₃C₂Tₓ@CoMo-LDH的Co K边XANES谱。(f) 异质结Co K边的FT-EXAFS谱。(g) CoMo-LDH、(h) Ti₃C₂Tₓ@CoMo-LDH异质结与(i) CoO中Co的k³加权小波变换K边EXAFS谱。
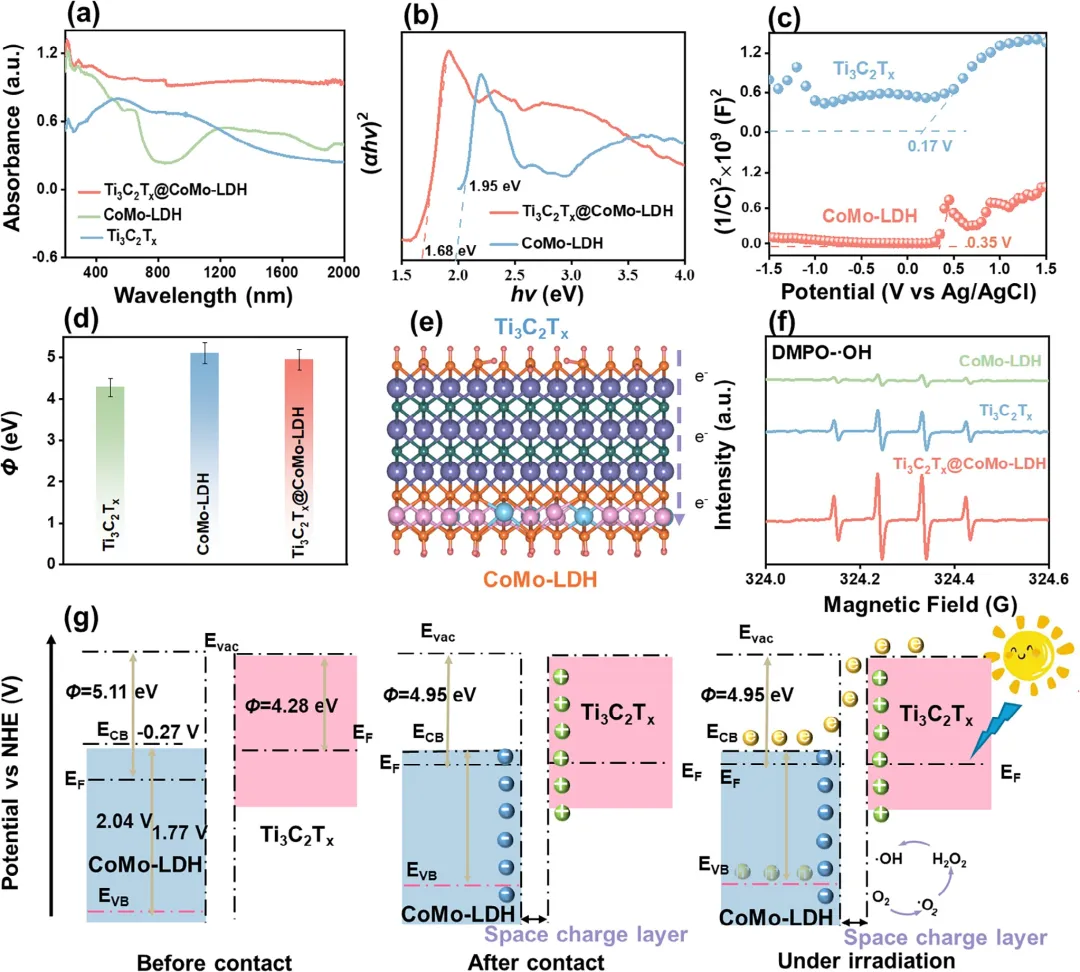
图4 (a)Ti₃C₂Tₓ、CoMo‑LDH和Ti₃C₂Tₓ@CoMo‑LDH的UV–vis-DRS光谱。(b) 利用Kubelka–Munk公式估算的CoMo‑LDH和Ti₃C₂Tₓ@CoMo‑LDH的带隙。(c) Ti₃C₂Tₓ和CoMo‑LDH的Mott–Schottky曲线。(d) 由CoMo‑LDH、Ti₃C₂Tₓ和Ti₃C₂Tₓ@CoMo‑LDH的UPS谱计算得出的功函数Φ。(e) Ti₃C₂Tₓ与CoMo‑LDH之间内建电场示意图。(f) Ti₃C₂Tₓ@CoMo‑LDH在太阳光照射下检测·OH的ESR谱图。(g) Ti₃C₂Tₓ@CoMo‑LDH肖特基异质结内的电荷转移路径。
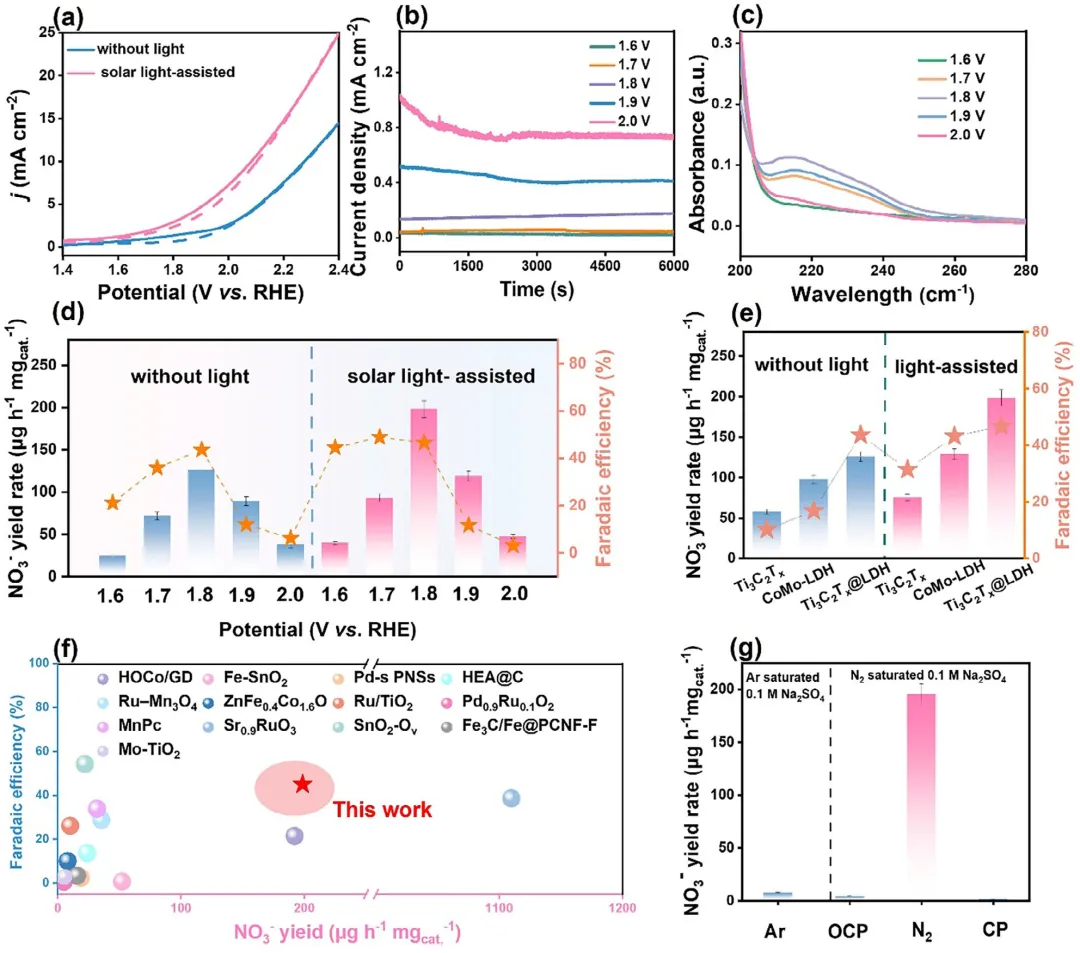
图5 (a) Ti₃C₂Tₓ@CoMo-LDH在N₂饱和(实线)与Ar饱和(点线)的0.1 M Na₂SO₄中,无光(蓝线)与模拟太阳光照(粉线)条件下的LSV极化曲线。(b) 模拟太阳光照下,不同外加电位eNOR测试后电解质的紫外-可见光谱。(c) 模拟太阳光照辅助下,Ti₃C₂Tₓ@CoMo-LDH在不同电位下的i–t曲线。(d) 暗态与光辅助条件下,Ti₃C₂Tₓ@CoMo-LDH在N₂饱和0.1 M Na₂SO₄中,6000 s内各外加电位下的NO₃⁻产率与FE关系。(e) 不同催化剂在1.8 V vs. RHE条件下,暗态与光辅助下的eNOR性能对比。(f) Ti₃C₂Tₓ@CoMo-LDH与其它已报道材料的eNOR活性比较。(g) 不同条件下碳纸(CP)与Ti₃C₂Tₓ@CoMo-LDH的eNOR性能。
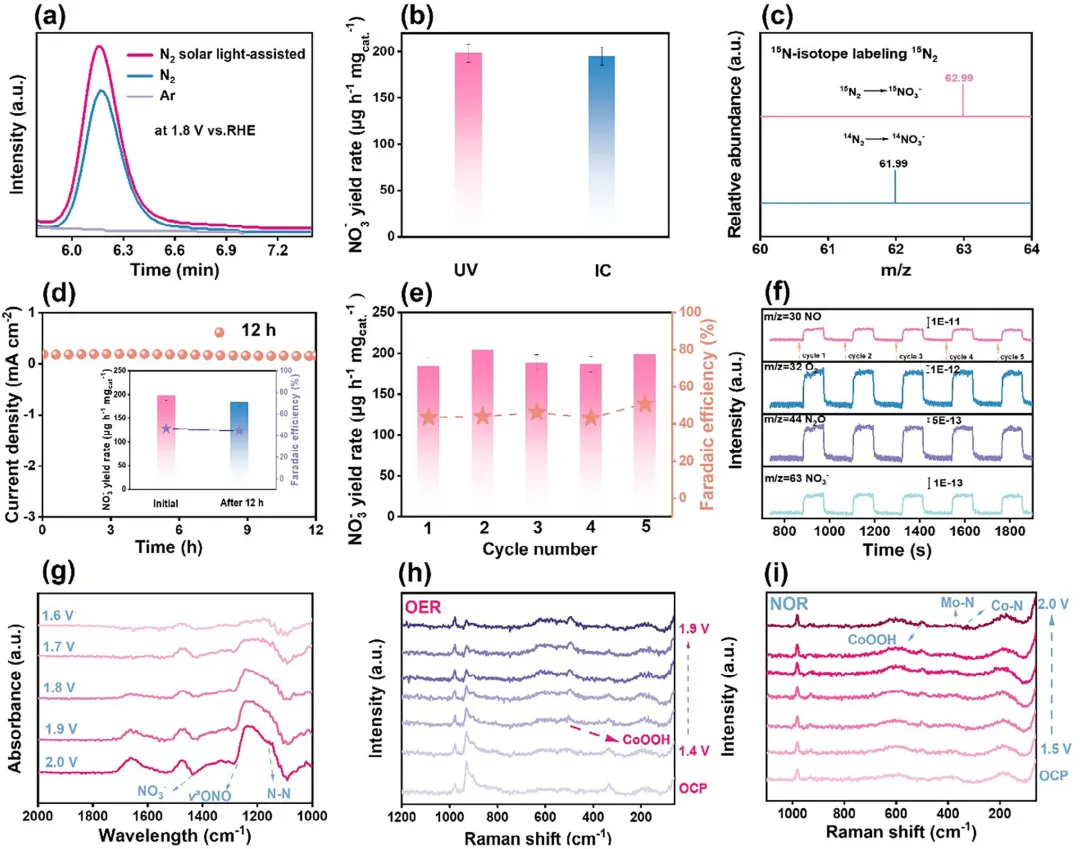
图6 (a) 产物的离子色谱图。(b) UV法与IC法测得的相应NO₃⁻产率。(c) ¹⁵N₂或¹⁴N₂气氛下NO₃⁻的质谱图。(d) 12小时计时电流测试(插图为原始与eNOR电解后的NO₃⁻产率与FE)。(e) Ti₃C₂Tₓ@CoMo-LDH在1.8 V vs. RHE下的循环稳定性。(f) Ti₃C₂Tₓ@CoMo-LDH在N₂饱和0.1 M Na₂SO₄电解质中,1.8 V vs. RHE下的在线DEMS测量。(g) Ti₃C₂Tₓ@CoMo-LDH在无N₂鼓泡的0.1 M Na₂SO₄中,不同电位下的原位FT-IR谱图。(h) Ti₃C₂Tₓ@CoMo-LDH在无N₂鼓泡的0.1 M Na₂SO₄中,不同电位下的原位拉曼谱图。(i) Ti₃C₂Tₓ@CoMo-LDH在N₂饱和0.1 M Na₂SO₄中,不同电位下的原位拉曼谱图。
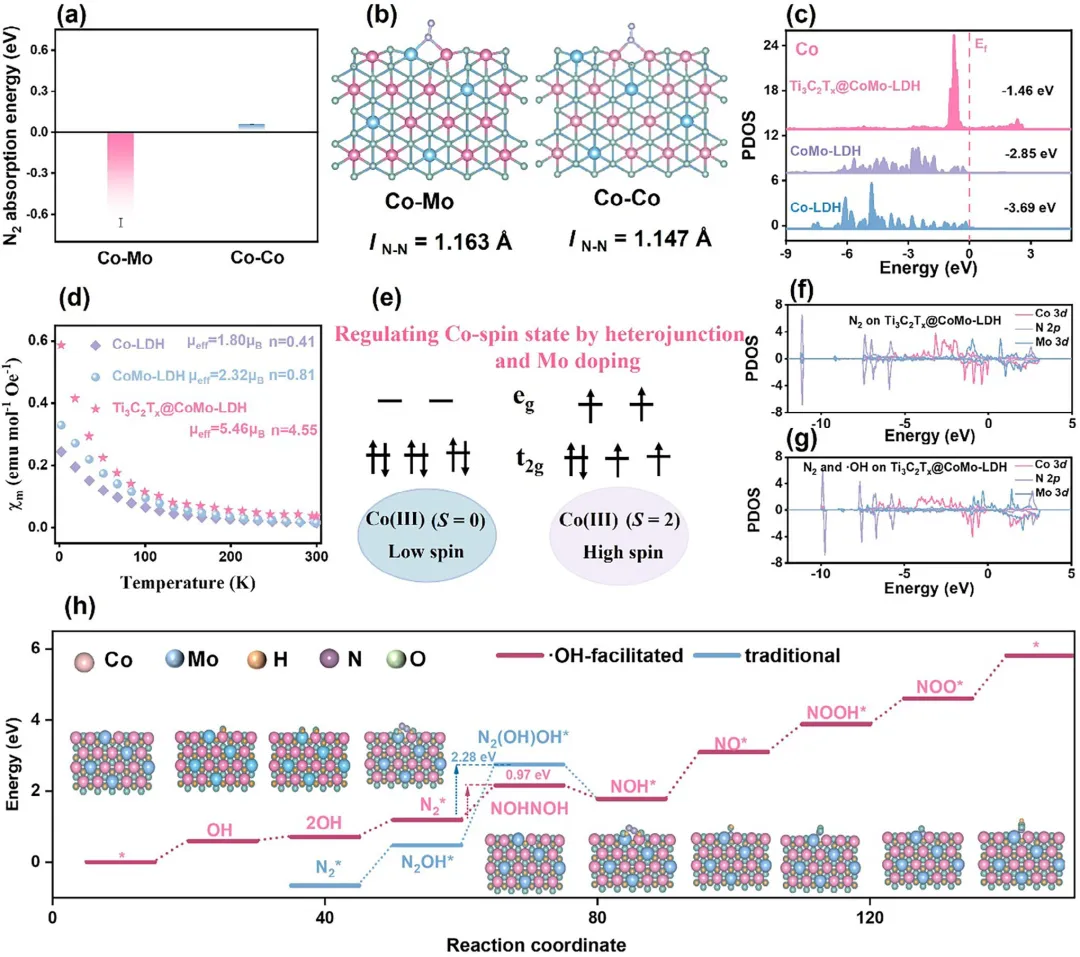
图7 (a) Co-Co位点与Co-Mo位点的N₂吸附能与(b)键长。(c) Co-LDH、CoMo-LDH与Ti₃C₂Tₓ@CoMo-LDH的Co轨道投影态密度PDOS。(d) Co-LDH、CoMo-LDH与Ti₃C₂Tₓ@CoMo-LDH的磁化率。(e) Ti₃C₂Tₓ@CoMo-LDH中Co(III)自旋态示意图。(f) Ti₃C₂Tₓ@CoMo-LDH上N₂吸附的PDOS。(g) ·OH促进下Ti₃C₂Tₓ@CoMo-LDH上N₂吸附的PDOS。(h) Ti₃C₂Tₓ@CoMo-LDH上不同路径eNOR的吉布斯自由能计算。
总之,为提升eNOR生产NO₃⁻的动力学与效率,该研究通过多策略协同构建了一种高效催化系统。为加速光生载流子的定向分离与迁移,通过构建Ti₃C₂Tₓ@CoMo-LDH n-n异质结引入了内建电场。此外,通过调控电子结构与Co³⁺自旋态,优化了Ti₃C₂Tₓ@CoMo-LDH中金属活性位点的电子云分布与轨道能级,从而改善了活性位点对N₂的吸附与活化及其与反应中间体*NOH的相容性。同时,Ti₃C₂Tₓ@CoMo-LDH利用模拟太阳光照下生成的大量·OH,为*NOH形成提供了充足的前驱体。这些多维调控策略协同作用,共同为增强eNOR反应动力学与NO₃⁻选择性生产奠定了坚实基础。此外,原位光谱表征揭示了电催化剂表面反应性CoOOH的形成,其中高自旋Co³⁺是驱动eNOR的实际活性位点。在模拟太阳光照下,光电Ti₃C₂Tₓ@CoMo-LDH展现出198.55 µg h⁻¹ mgcat⁻¹的高NO₃⁻产率与46.22%的FE,超越了大多数过渡金属催化剂。该研究提出了一种基于固氮作用、通过eNOR将N₂转化为NO₃⁻的简短反应路径,为NO₃⁻电合成提供了新途径。