突破常规!郑州大学陈学年《Nat. Catl.》钯催化反迈克尔型不对称加成,解锁碳硼烷高效偶联
点蓝色字 关注“化解 Chem”
关注“化解 Chem”
第一作者:Chao Lei, Wen Lu, Tingting shen
通讯作者:Donghui Wei, Yan-Na Ma, Xuenian Chen
通讯单位:Zhengzhou University文章链接:https://doi.org/10.1038/s41929-026-01480-4
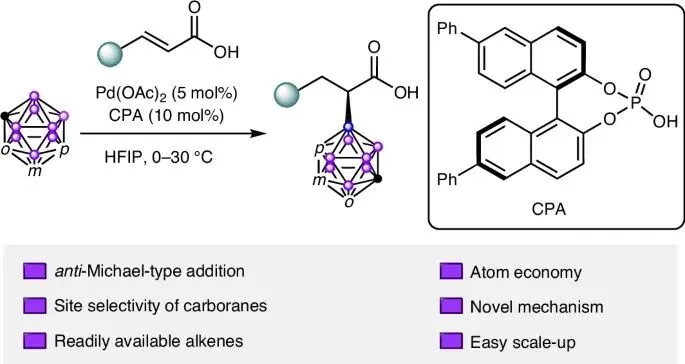
今天,分享一个钯催化反迈克尔型不对称加成研究成果,该文章发表在《Nature Catalysis》期刊该研究报道了一种新颖的钯催化不对称反应,实现了亲核试剂(碳硼烷)对α,β-不饱和羧酸的反向α-加成,突破了传统的迈克尔加成模式。该方法具有高选择性、高效性和广泛适用性,为合成手性α-取代羧酸提供了新策略,并通过机理研究揭示了其选择性来源。该研究领域的背景、面临的挑战以及本研究提出的创新性解决方案:研究背景与动机:
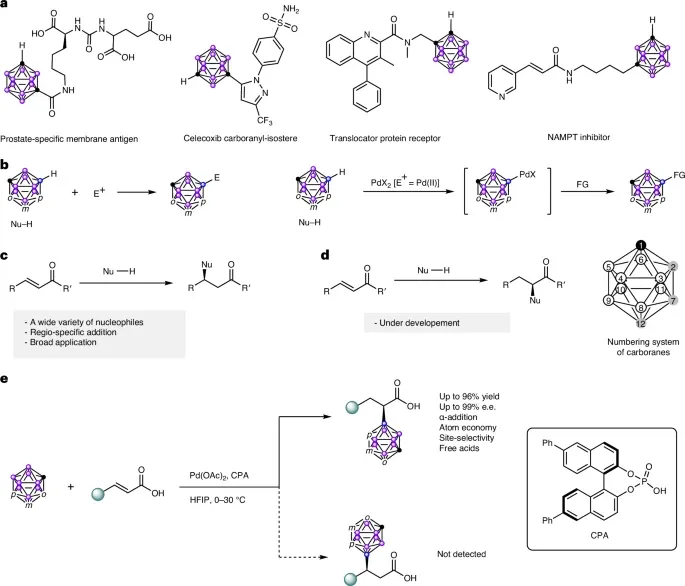
- 二十面体碳硼烷(即C2B10H12)是一类具有三维芳香性的代表性硼簇合物,在材料化学、有机合成和药物领域具有潜在应用。特别是在药物化学中,碳硼烷衍生物因其能够形成非常规的二氢键而备受关注,这种键可以在设计生物活性化合物时靶向不同生物受体的疏水结合位点
- 催化不对称迈克尔加成反应因其原子经济性和环保特性,是构建α,β-不饱和羰基化合物β-手性中心最有价值的方法之一
面临的合成挑战:
- 由于B−H键的弱极性以及邻/间碳硼烷中十个B−H键的低选择性,笼硼取代的邻碳硼烷衍生物相对较少
- 将硼顶点与手性碳中心连接的催化不对称合成方法此前尚未见报道
- 催化不对称迈克尔加成反应因其原子经济性和环保特性,是构建α,β-不饱和羰基化合物β-手性中心最有价值的方法之一
本研究的创新策略:
设想碳硼烷是否可以作为一类迈克尔供体与α,β-不饱和羰基化合物反应。然而,碳硼烷的低亲核性使其无法直接进行亲核加成
硬亲电试剂倾向于与苯反应,而软亲电试剂则明显偏好邻碳硼烷
作为一种软亲电试剂(路易斯酸),Pd(II)对邻碳硼烷表现出高反应活性,并能在温和条件下活化B−H键
基于实验和计算结果,研究了反应机理和对映选择性的来源,揭示了稳定的五元钯环中间体的形成是实现反向选择性的关键
选择了间碳硼烷作为模型底物,因为间碳硼烷与邻碳硼烷表现出相似的反应活性
Pd(II)催化剂对邻碳硼烷显示出高反应活性,能够通过亲电钯化过程形成B-Pd中间体,从而在温和反应条件下活化B-H键
HFIP溶剂在邻/间碳硼烷的选择性B(9)-H活化中发挥着不可替代的作用
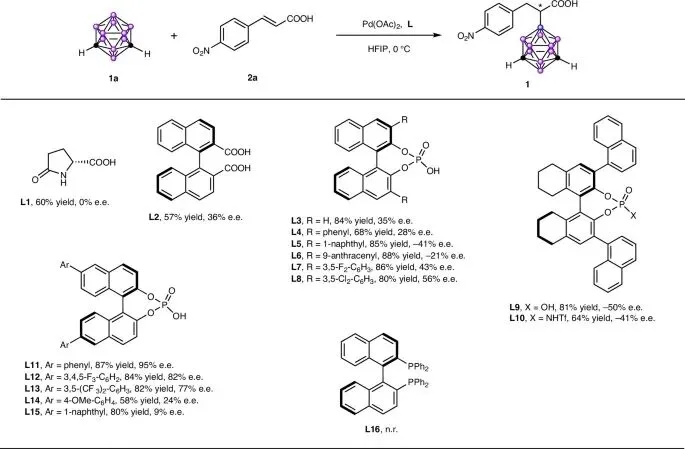
探索了多种布朗斯特酸,包括手性氨基酸、羧酸和磷酸
使用手性联萘二甲酸L2和联萘酚磷酸酯L3作为配体时,检测到了36%和35%的对映体过量,且使用L3时获得了高产率
在模型反应中测试了一系列手性磷酸衍生物L4-L15,结果表明,在6,6'-位带有芳基取代基的相应BINOL衍生手性磷酸对实现立体控制至关重要。其中,配体6,6'-二苯基-联萘酚磷酸酯L11给出了最佳结果(产率87%,对映体过量95%)。
图片来源:Natrue Synthesis
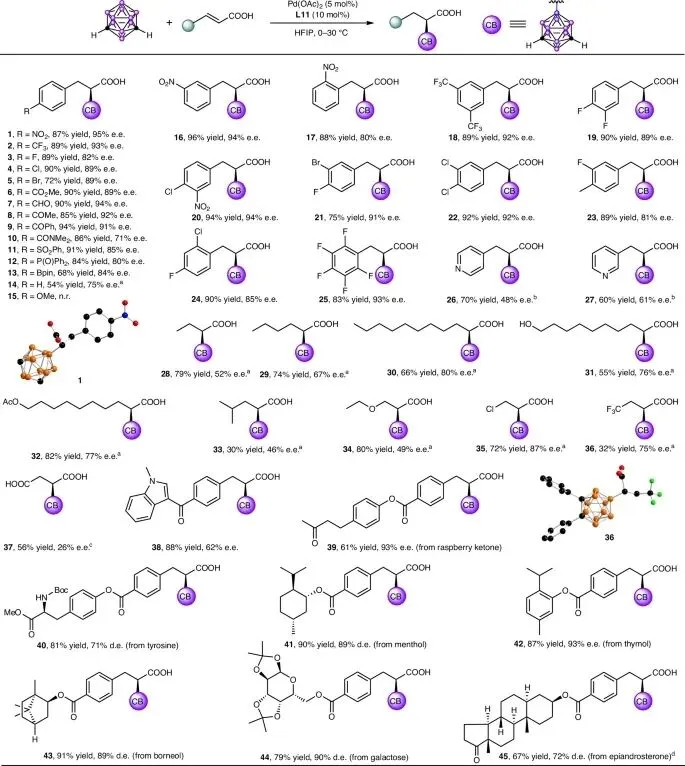
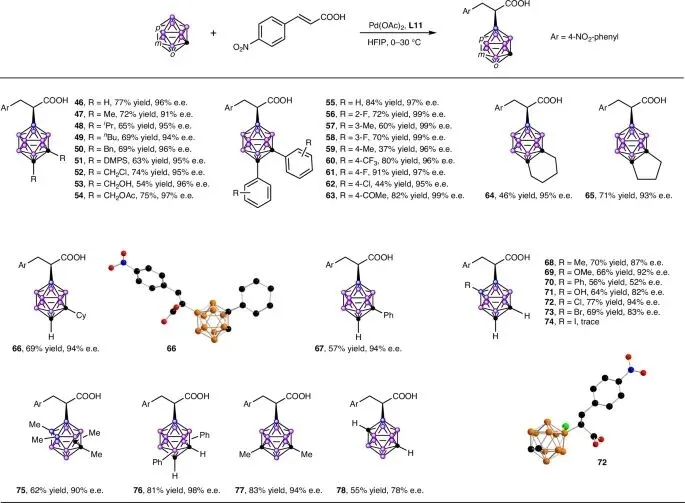
系统考察了α,β-不饱和羧酸的结构多样性对钯催化不对称反迈克尔型加成反应的影响。实验结果表明,该反应具有广泛的底物适用性和良好的官能团耐受性:
芳基取代羧酸:多种带有不同取代基(对位、间位、邻位)的肉桂酸衍生物均可顺利反应,以中等到优异的产率(71-95% e.e.)得到目标产物。缺电子基团取代的底物表现更佳,而强给电子基团(如甲氧基)则导致反应无法进行。值得注意的是,反应兼容硼酸酯和醛基等敏感官能团。
杂芳基与烷基取代羧酸:含氮杂环底物因氮原子的强配位作用抑制反应,但加入三氟甲磺酸(HOTf)保护氮原子后,可成功获得中等产率和ee值的目标产物。β-烷基取代的丙烯酸类底物中,长链烷基有利于提高对映选择性;而大位阻的异丙基取代底物则产率和ee值较低。
β-位缺电子取代羧酸:当β-位为乙氧基、氯原子等缺电子基团时,反应仍能成功在α-位引入碳硼烷基。三氟甲基取代底物则得到α-和β-位加成产物的混合物,其中α-位产物为主,其绝对构型通过X射线单晶衍射得以确认。
生物活性分子后期修饰:该方法展现出优异的官能团兼容性,成功应用于吲哚、树莓酮、表雄酮、酪氨酸、薄荷醇、麝香草酚、冰片、半乳糖等多种生物活性分子和天然产物的后期修饰,以良好至优异的产率(62-93% e.e.)得到相应目标产物,为手性含硼药物分子的快速构建提供了新途径。
碳硼烷的适用性:
图片来源:Natrue Synthesis
结果表明该催化体系对多种碳硼烷衍生物均表现出良好的适用性和优异的对映选择性控制:
邻碳硼烷及其C-取代衍生物:与间碳硼烷类似,邻碳硼烷顺利反应得到目标产物46,产率77%,ee值96%,并表现出良好的B(9)区域选择性(B9/B8=9:1)。一系列双C-取代的邻碳硼烷(包括烷基、芳基、硅基等)均能兼容,以中等到良好的产率(37-91%)和优异的对映选择性(91-99%)得到相应产物。值得注意的是,硅基在该条件下保持完整,避免了传统Pd(II)催化中常见的消除副反应。单C-取代邻碳硼烷反应得到B(8)、B(9)、B(10)、B(12)位产物的混合物,但主要分离得到B(9)位产物,且对映选择性优异(化合物66的绝对构型经X射线单晶衍射确证)。
B-取代邻碳硼烷:硼顶点上的取代基对反应产率和对映选择性有显著影响。例如,9-苯基取代的邻碳硼烷导致产物70的ee值降至52%,而9-碘代底物则完全无效。B(9)和B(12)双取代的底物反应发生在B(8)位,获得产物75;B(3,6)位取代的底物则表现良好,高产率高对映选择性得到产物76。
间/对碳硼烷:1,7-二甲基间碳硼烷顺利反应,得到产物77(83%产率,94% ee)。惰性更高的对碳硼烷在该体系中也能够反应,以55%产率和78% ee得到目标产物78。
通过该方法合成的产物可作为多功能的合成砌块,进行多样化的后期衍生:图片来源:Natrue Catalysis
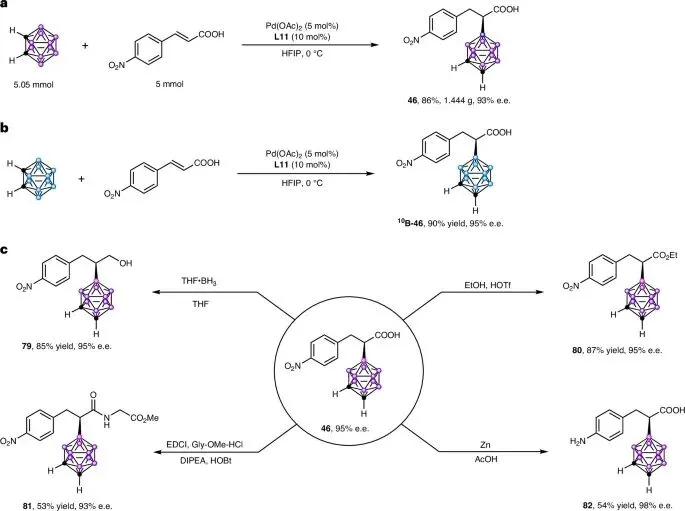
规模化与同位素标记:邻碳硼烷与4-硝基肉桂酸的克级规模反应顺利获得产物46,产率86%,ee值93%,展示了反应的稳健性。该方法同样适用于1010B富集的邻碳硼烷,高效合成硼中子俘获治疗所需的1010B标记手性分子1010B-46(90%产率,95% ee)。
官能团衍生化:产物46中的羧基和硝基可进行多样化转化,且手性中心在酸/碱条件下保持稳定。具体包括:羧基还原为醇(79,85%产率,ee值不变)、酯化(80,87%产率,ee值不变)、与甘氨酸甲酯缩合形成酰胺(81,53%产率,93% ee),以及硝基选择性还原为氨基(82,54%产率,98% ee)。
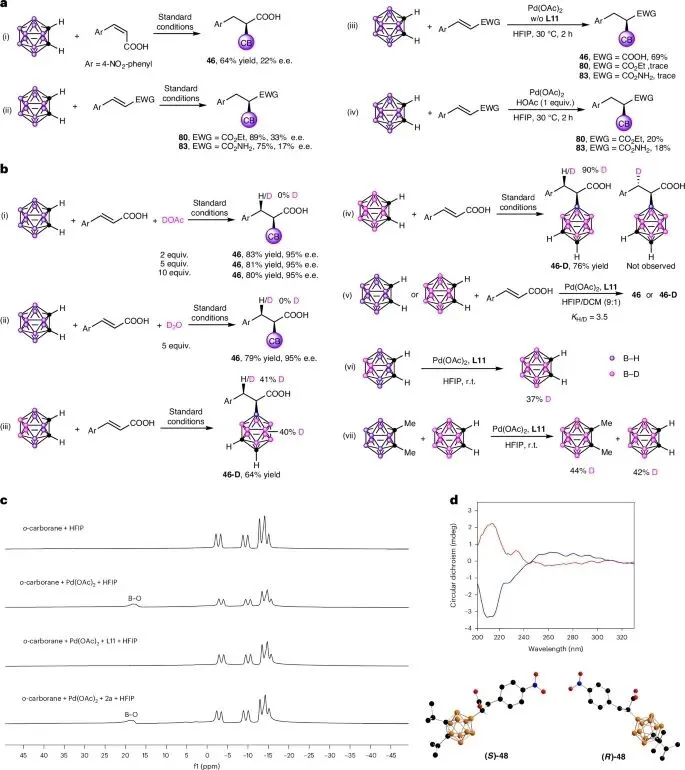
构型与酸性质子必要性:反应需(E)-构型烯酸以获得高对映选择性;酸性α,β-不饱和羧酸是必需底物,其酯或酰胺在标准条件下仅得痕量产物且ee值低,但外加乙酸可促进反应,证实羧基质子关键作用。
氢来源与B-H断裂决速:氘标实验证明产物β-H完全源于碳硼烷B-H,而非溶剂或外加酸;平行动力学实验KIE=3.5,表明B-H键断裂为决速步。
顺式加成与钯迁移:氘代碳硼烷实验表明,B-D通过顺式加成转移到烯酸β位;反应中观测到碳硼烷硼顶点间的H/D均一化分布,表明存在可逆的钯迁移过程。
手性磷酸的双重作用:11B NMR监测显示,无手性磷酸时Pd(II)会促使碳硼烷快速分解为B-O副产物;加入手性磷酸后,通过形成稳定的B-Pd-阴离子物种抑制分解,从而导向目标催化循环。
对映体确证:通过圆二色光谱验证了由不同构型手性磷酸配体得到的产物46具有相反的绝对构型,互为对映体;同时利用X射线单晶衍射确证了产物48的绝对构型,为立体化学指认提供了直接证据。
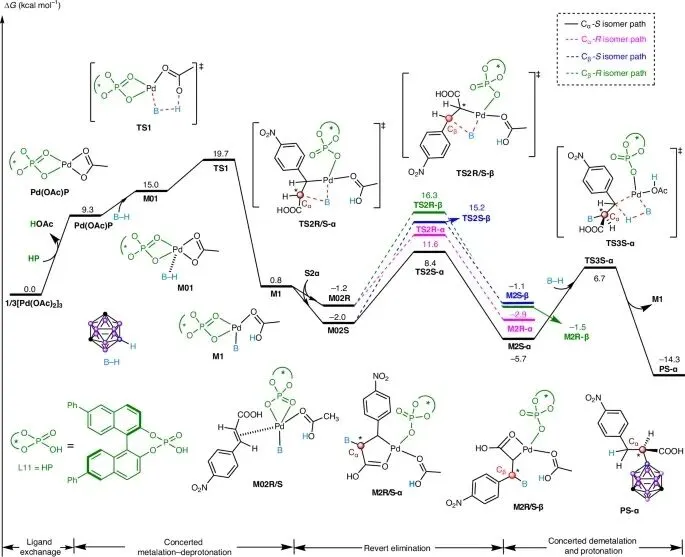
DFT计算确认CMD机理:理论计算表明,反应通过CMD机理进行,其中B-H键活化(TS1,能垒19.7 kcal mol⁻¹)为决速步。相比之下,协同氢负离子转移路径(能垒>43 kcal mol⁻¹)和B-H插入形成Pd-H物种的路径(能垒25.4 kcal mol⁻¹)能量上均不利,因此被排除。
α-选择性与S-构型偏好:计算显示,α-加成路径的能垒(10.4-12.8 kcal mol⁻¹)显著低于β-加成路径(17.2-17.5 kcal mol⁻¹)。在α-加成路径中,生成S-构型产物的过渡态能垒(10.4 kcal mol⁻¹)低于生成R-构型产物(12.8 kcal mol⁻¹),与实验观察到的主要生成S-构型产物的结果高度吻合。
动力学研究验证:反应动力学测定显示对碳硼烷、手性磷酸和钯催化剂为一级,对不饱和羧酸为逆级数。这证实了碳硼烷、催化剂和手性磷酸参与决速步,而不饱和羧酸的逆级数归因于其阴离子配位后形成的催化剂Pd(S₂)L11活性低于原乙酸根配位的Pd(OAc)L11。
CMD机理的实验佐证:当使用Pd(TFA)₂代替Pd(OAc)₂时反应几乎不进行,但外加乙酸钠等碱后可恢复反应活性并获得中等结果。这直接证实了B-H活化需要碱(乙酸盐)参与,符合CMD机理特征,并说明乙酸盐配位的Pd(OAc)L11是更优的活性催化物种。
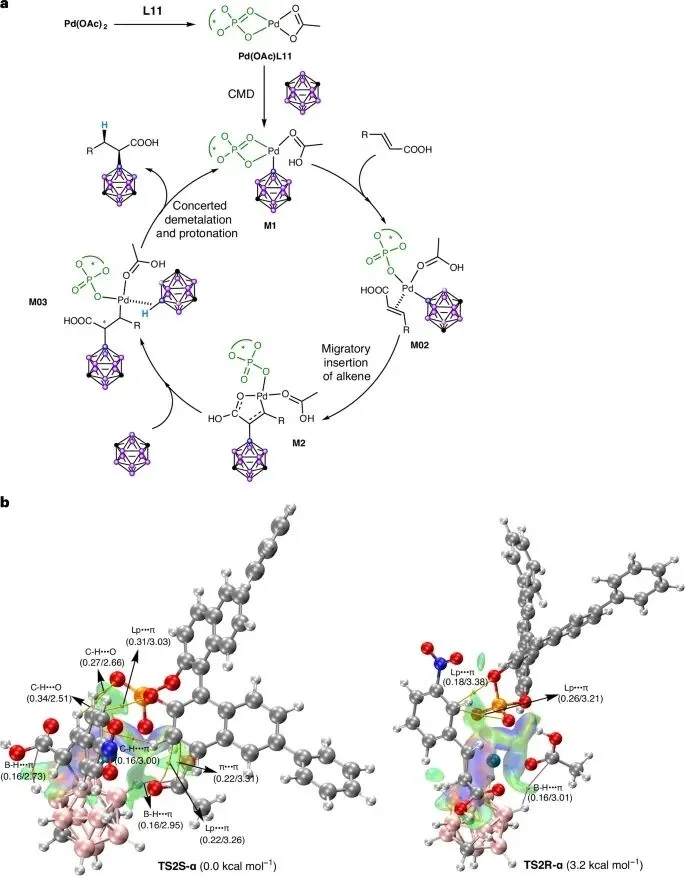
反应机理如下图所示:
图片来源:Natrue Catalysis
催化循环:反应始于手性催化剂Pd(OAc)L11的生成,随后经历CMD机理的B-H活化、烯烃配位与迁移插入(形成关键的五元钯环中间体M2),最后经协同脱金属/质子化释放产物并再生催化剂。其中,五元钯环M2的形成是控制α-区域选择性的关键。
立体选择性来源:通过对决定手性的两个关键过渡态TS2S-α(生成S-产物)和TS2R-α(生成R-产物)进行非共价相互作用分析,揭示了S-选择性的起源。
弱相互作用差异:分析表明,TS2S-α中存在着比TS2R-α中更强且更多的弱相互作用,包括B−H···π相互作用和孤对电子···π相互作用。特别是,在TS2S-α中,烯烃底物与反应体系之间存在多种弱相互作用(如C−H···π、π···π和C−H···O),而在TS2R-α中这些相互作用基本缺失。
能量差异:TS2S-α中更丰富、更强的弱相互作用网络有效稳定了该过渡态,使其能垒低于TS2R-α,从而在能量上更有利,最终决定了反应的对映选择性偏好(主要生成S-构型产物)。
本研究报告了一种钯催化α,β-不饱和羧酸与碳硼烷的不对称反迈克尔型加成反应。该反应以手性磷酸为配体,在温和条件下实现了邻、间、对碳硼烷在羧酸α-手性中心的高效、高选择性引入。肉桂酸及其衍生物、烷基取代丙烯酸等一系列底物均适用,以高产率和高对映选择性得到α-碳硼烷基羧酸产物。结合实验与计算化学研究,阐明了反应机理和对映选择性的起源——关键的五元钯环中间体决定了α-区域选择性,而过渡态中差异化的弱相互作用网络(如B−H···π、孤对电子···π等)则控制了立体选择性。该方法为合成手性α-碳硼烷基羧酸提供了简洁通用的途径,目前正在进一步拓展亲核试剂与烯烃底物的适用范围。陈学年,二级教授,博士生导师,郑州大学学科特聘教授,河南省硼化学与先进能源材料重点实验室主任,河南省硼化学国际联合实验室主任,河南省化学会常务理事。长期从事硼化学,金属有机化学,无机化学,配位化学和材料化学等领域研究。在硼化学、储氢材料、特种功能材料等领域有一定学术影响。2013年回国后主持一项国家自然科学基金-河南联合重点基金和三项国家自然科学基金面上项目;在美国工作期间主持或参与一项美国能源部、两项美国自然科学基金和一项横向研究项目。在Acc. Chem. Res., J. Am. Chem. Soc., Angew. Chem. Int. Ed., Nat. Commun., Chem. Sci.等国际知名学术杂志发表学术论文100余篇,拥有多项美国和中国专利技术。多次应邀在国际、国内会议和著名高校、科研院所做邀请报告。研究方向:硼化学,有机化学,金属有机化学,配位化学
主持项目:1.硼氮化合物中的双氢键及其诱导氢气生成反应的影响因素及一般规律研究(21371051,2014.01-2017.12)
2.饱和硼氮烃类似物的可控合成和反应机理研究
(21771057,2018.01-2021.12)
3.新型硼基固体离子导体材料的制备及其导电机理研究(U1804253,2019.01-2022.12)
4.利用硼氢键缩合反应高效合成多面体硼烷化合物及机理(22171246,2022.01-2025.12)


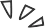
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
