郑州大学卢思宇/常江伟,最新Angew!定制化O-O自由基耦合路径助力高密度Fe-N-C催化剂实现稳定工业规模水氧化!胡志昂一作
丨旨在分享学术交流丨能力有限欢迎指正丨
「上海岱算科技有限公司」已向境内外230余家高等院校/科研院所提供了累计1400多项模拟计算服务,赋能科学研究提速增效!合作实验课题组在线发表学术论文期刊有ACS系列、AM系列、Angew、CEJ、EST、JACS、Matter、Nature子刊等,助力科研工作锦上添花!低成本、高效且耐用析氧反应(OER)催化剂对于通过阴离子交换膜水电解推进大规模制氢至关重要,然而当前最先进的催化剂受限于在高电流密度下运行稳定性不足,以及在动态操作条件下缺乏对反应路径的精确控制。
2026年06月03日,郑州大学卢思宇、常江伟团队在Angewandte Chemie International Edition期刊发表题为“Customized O-O Radical Coupling Route on High-Density Fe-N-C Catalysts for Stable Industrial-Scale Water Oxidation”的研究论文,团队成员胡志昂为论文第一作者,卢思宇、常江伟为论文共同通讯作者。
第一作者:胡志昂
通讯作者:卢思宇、常江伟
通讯单位:郑州大学
论文DOI:10.1002/anie.9504269
该研究通过理论计算揭示了Fe位点密度对Fe-N-C单原子催化剂上关键OER中间体吸附能的连续调控效应。实验结果进一步表明,通过逐步增加Fe位点密度,可以对表面*OH覆盖度进行精细调控,从而实现OER机制从吸附质演化机制到氧化物路径机制(OPM)的可控转换。遵循OPM的高密度Fe-N-C催化剂(HDFe-N-C,Fe负载量为14.7wt.%)表现出卓越的电化学稳定性,在500mAcm⁻²的电流密度下以288mV的低过电位连续稳定运行2000小时。当集成到阴离子交换膜电解槽中时,该器件在仅2.15V电压下即可达到5Acm⁻²的电流密度,并维持稳定性能500小时。
阴离子交换膜水电解(AEMWE)已成为一种在低温下高效制氢的有前景的技术,它结合了碱性水电解(AWE)的成本效益和高能效,以及质子交换膜水电解(PEMWE)的高电流密度能力和高氢气纯度的特点。然而,AEMWE技术的实际应用面临重大挑战,特别是在高电流密度(≥5Acm⁻²)下,缓慢的析氧反应(OER)动力学和不足的长期器件稳定性严重限制了其性能和规模化部署。最先进的系统通常限制在5Acm⁻²以下运行,而基于传统材料的AEMWE器件通常仅能维持数小时的操作寿命。这种性能限制可能解释了为何关注高电流密度操作条件的研究报道数量有限。因此,开发合成简便、成本效益高、且能够在安培级电流密度下提供高OER活性和出色稳定性的OER电催化剂,对于推动AEMWE技术的工业化进程至关重要。
单原子催化剂(SACs)结合了均相和多相催化的结构和功能优势,由于其不饱和配位环境、接近100%的金属原子利用效率以及明确的活性位点,在水分解电催化领域展现出卓越前景。这些特性使SACs成为实现高效电化学能量转换的理想候选材料。然而,在实际器件制备的背景下,M-N-C SACs通常较低的金属负载量是一项关键瓶颈。为了达到足够的催化活性,通常需要较厚的催化层,这会显著加剧传质阻力,导致过电位增加和稳定性下降,从而严重损害器件性能。因此,基础研究进展与工业应用之间仍然存在巨大差距。此外,金属原子的稀疏分布导致空间上孤立的活性位点,在OER过程中强制执行固定的吸附质演化机制(AEM),其中含氧中间体在单个原子位点上依次吸附和转化。该过程受强线性标度关系制约,这阻碍了对不同中间体结合能的独立优化,并从根本上限制了对使用替代OER路径的更高效OER催化剂的理性设计。因此,关于SACs在AEMWE中应用的报道仍然很少,特别是在≥5Acm⁻²的工业级操作条件下,相关研究进展几乎空白。
因此,具有高原子位点密度(即高负载量)的SACs的理性设计与合成具有重大的基础研究意义和实际应用价值。随着金属负载量的增加,传统的电催化机制已不足以完全解释SACs的催化行为,人们越来越认识到非常规反应路径的存在。孤立活性位点所带来的限制不再严格维持;相反,高密度金属中心可以参与协同相互作用,产生非常规催化机制并偏离经典的标度关系,从而显著调节电化学活性。其中,氧化物路径机制(OPM)是最近在SACs中提出的一种非常规机制,其通过相邻*O中间体的直接耦合实现O-O键的形成,与吸附质演化机制相比提供了更优异的OER活性。值得注意的是,OPM不涉及晶格氧的参与,这使其区别于晶格氧机制(LOM),从而规避了由晶格氧迁移导致的结构退化以及因金属浸出引起的催化剂快速失活。在三维过渡金属氧化物中,杂原子掺杂水平和刚性的体相结构限制了表面灵活性。因此,氧化物表面通常无法满足有效*O→*O耦合所需的局部几何构型。相比之下,负载在二维氮掺杂碳材料上的SACs受益于增强的结构灵活性和可调性,能够实现有利于*O→*O耦合的最佳空间排布。这种固有优势促进了当金属负载量足够高时,SACs体系中OPM的优先活化,有效打破了传统标度关系的限制,降低了反应能垒,并加速了OER动力学。从机理角度来看,能够合成具有宽范围金属负载量的SACs是非常理想的,因为这可以精确调控局域配位环境和金属位点间距,从而实现多金属协同、电子结构演化和反应路径选择,为深入阐明OPM的化学和结构起源提供了一个理想的模型平台。
在此,该研究首次报道了一种在AEMWE操作条件下表现出卓越稳定性的高密度铁单原子催化剂(HDFe-N-C)。通过制备具有宽范围(高达~15wt.%)Fe负载量的Fe-N-C催化剂,实现了对表面*OH覆盖度的精确和连续调控,从而能够将OER路径从低Fe负载量时的AEM可控地切换到高Fe负载量时的OPM。结合理论和实验证据,该研究证明高金属密度的HDFe-N-C催化剂通过OPM路径,经由两个相邻*O中间体的直接耦合促进O₂的形成,提供了卓越的OER性能:在500mAcm⁻²的电流密度下以288mV的低过电位稳定运行2000小时。更重要的是,HDFe-N-C在AEMWE系统中,于5Acm⁻²的工业级电流密度下保持了500小时的优异稳定性。此外,在0~5Acm⁻²之间进行3500次快速启动-停止循环后,未观察到可测量的性能衰减。该研究不仅展示了一种能够在严苛的动态和高电流密度条件下持续进行OER的稳健单原子催化剂,突显了其巨大的实际应用潜力,还通过阐明活性位点结构演化与OPM路径激活之间的内在关联,为高效OPM型催化剂的理性设计提供了基础性见解。
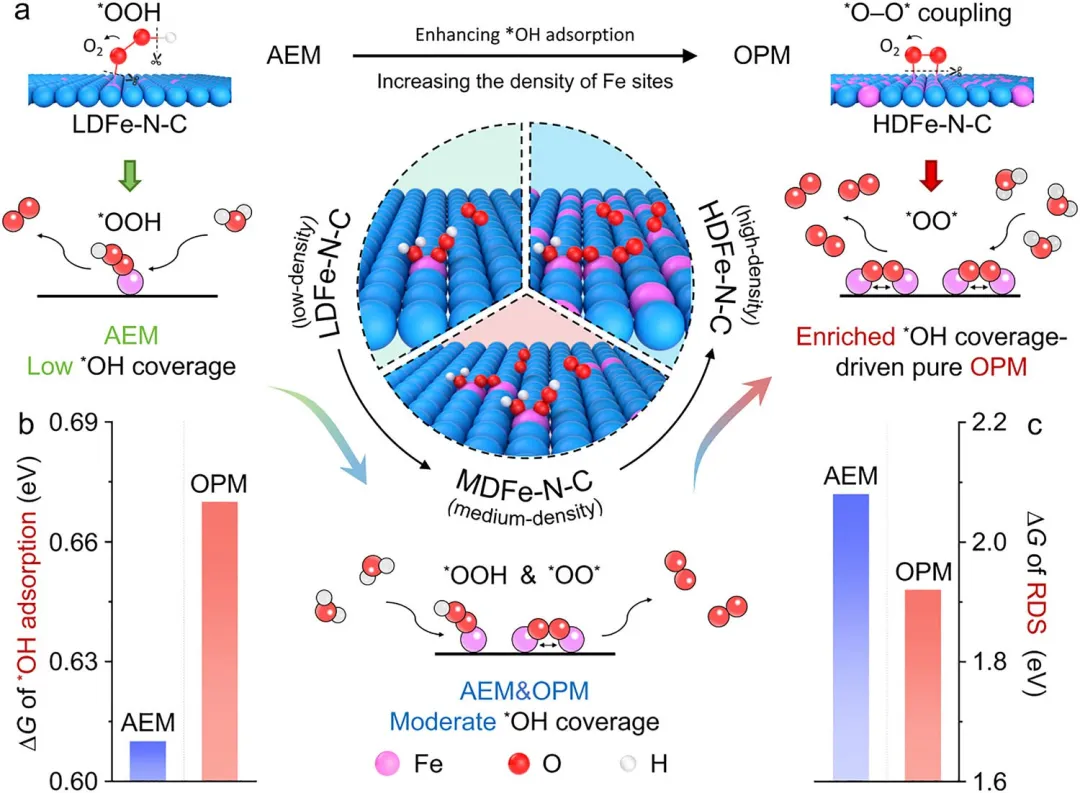
图1 | 不同Fe位点密度下OER反应机制。(a) 通过金属密度调控OER路径的示意图。HDFe-N-C表面促进*OH的富集,从而实现OPM的触发。(b) HDFe-N-C和LDFe-N-C上*OH吸附的相对能量。(c) HDFe-N-C和LDFe-N-C上遵循AEM和OPM路径的ΔG图比较。基于OPM的HDFe-N-C表现出降低的反应能垒。
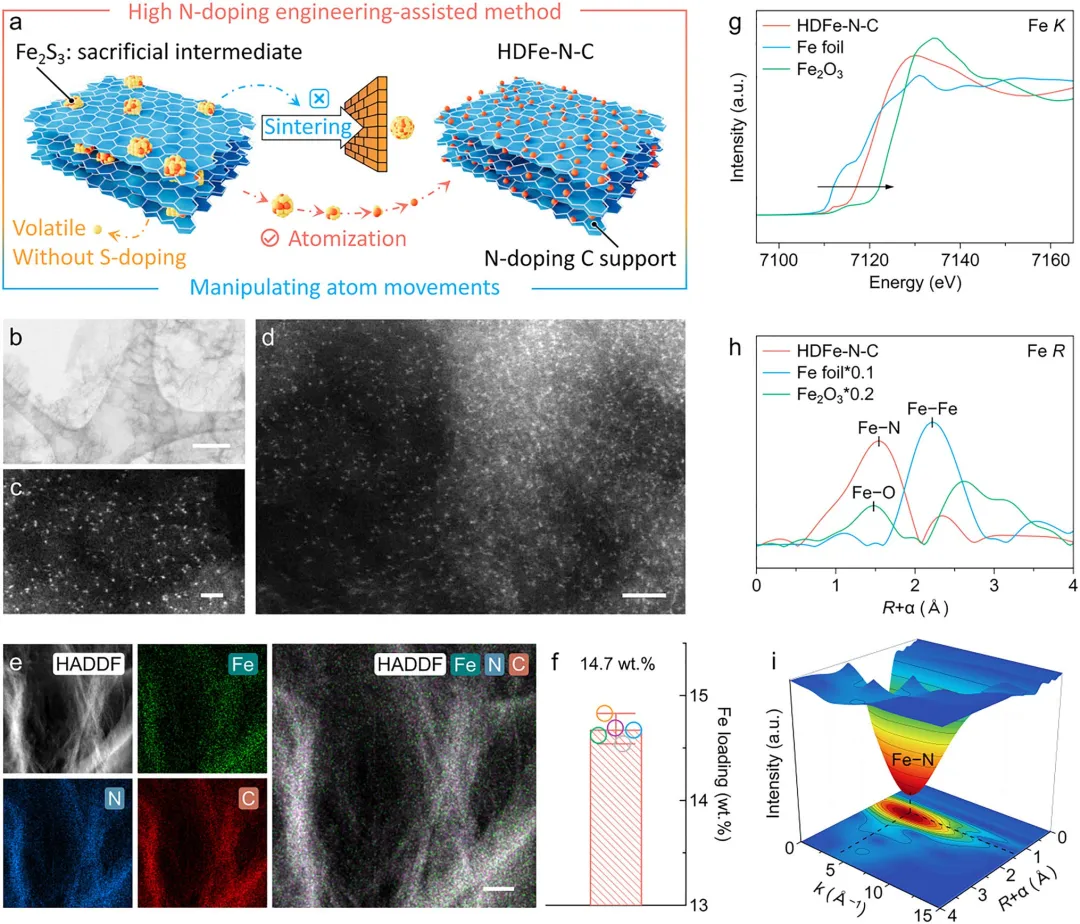
图2 | HDFe-N-C的原子结构和元素分布。(a) 高氮掺杂工程辅助的HDFe-N-C合成策略。(b-d) AC-HAADF STEM图像显示HDFe-N-C中高度分散且孤立的Fe原子。(e) HDFe-N-C的ADF图像以及Fe、N、C的局部EDX元素分布图及其叠加图像。(f) 通过ICP-OES测定的HDFe-N-C中的平均Fe掺杂含量。(g) HDFe-N-C及选定参考材料(包括Fe₂O₃和Fe箔)的归一化Fe K-edge XANES谱图。(h) HDFe-N-C、商用Fe₂O₃和Fe箔的归一化FT Fe K-edge EXAFS谱图。(i) HDFe-N-C的k³加权WT-EXAFS等高线图。图(b)、(c)、(d)和(e)中的比例尺分别为200、1、2和100nm。
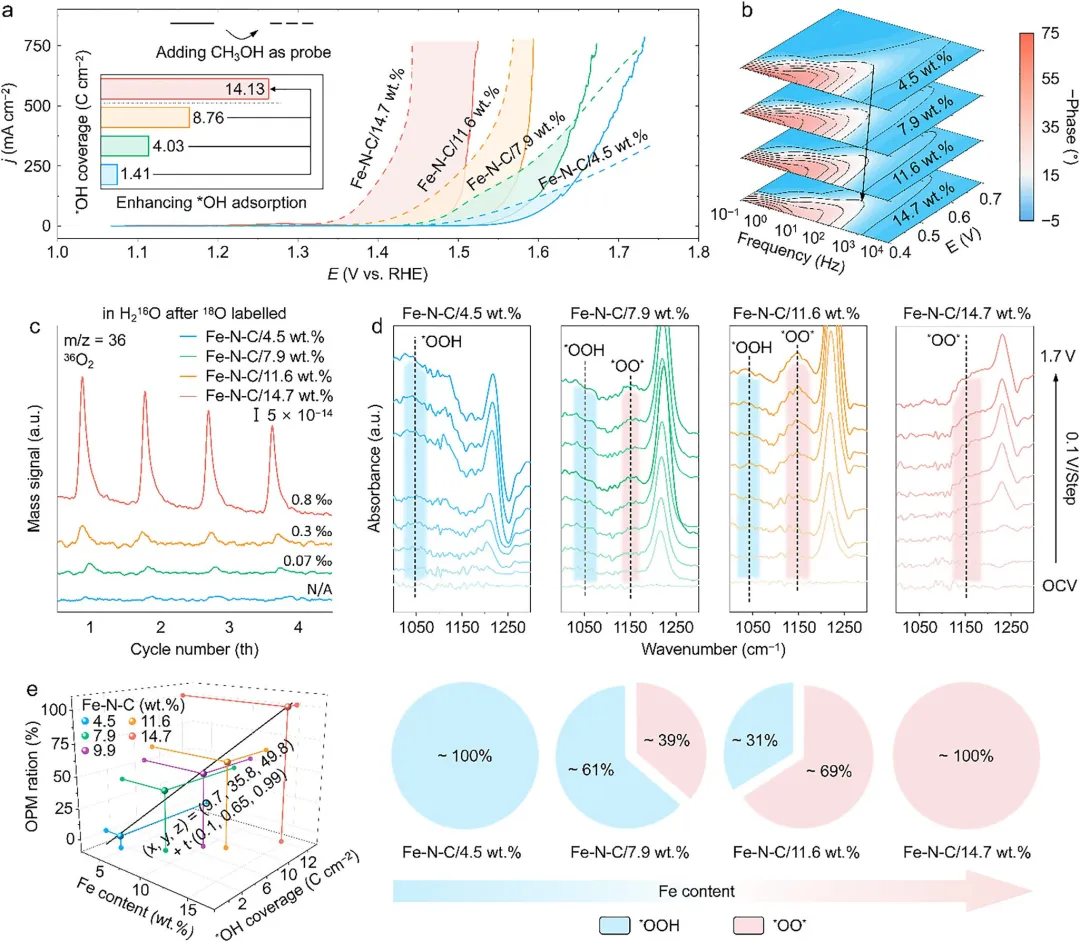
图3 | 探索具有不同Fe负载量的Fe-N-C催化剂上的OER机制。(a) OER和MOR曲线。插图为极化曲线的电流差值。(b) operando电化学阻抗谱。(c) 从operando DEMS获得的³⁶O₂信号的详细比较。(d) operando ATR-SEIRAS谱图(上图)以及位于~1025和~1150cm⁻¹的特征峰强度比率变化(下图)。(e) 对于具有梯度Fe负载量的Fe-N-C催化剂,Fe含量(x轴)、*OH覆盖度(y轴)和OPM比例(z轴)之间的定量3D相关图。
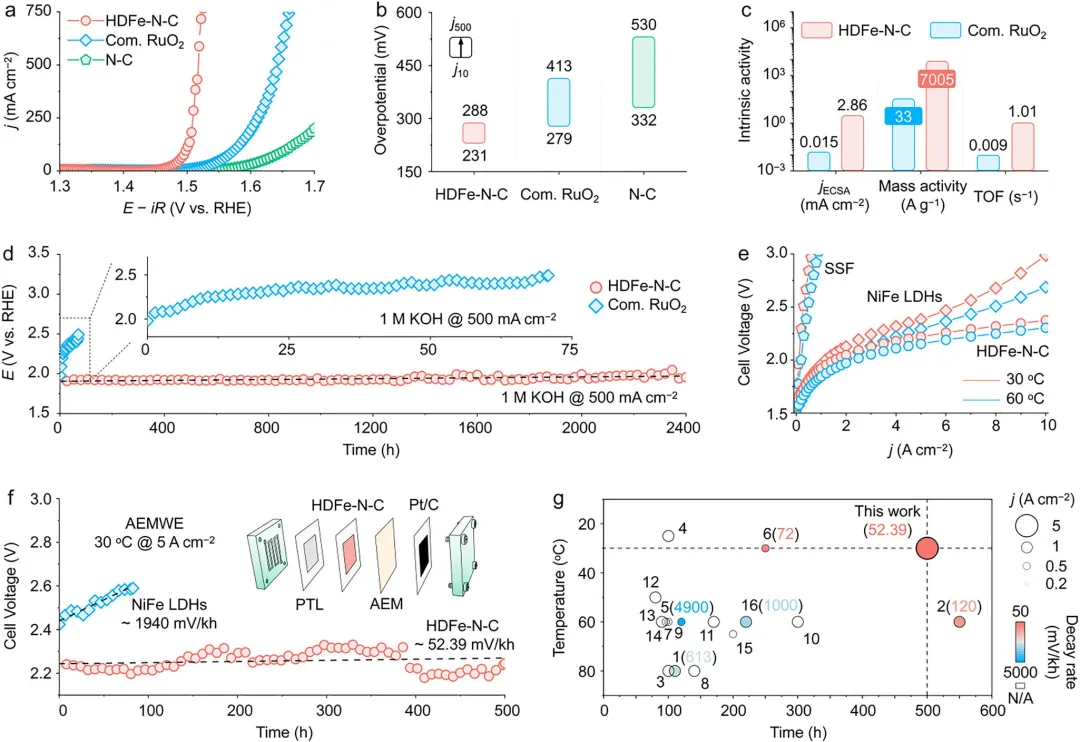
图4 | HDFe-N-C催化剂的OER性能。(a) HDFe-N-C、商用RuO₂和N-C在三电极体系中经iR校正(95%)后的OER极化曲线,以及HDFe-N-C在快速循环伏安加速老化测试中伴随扫描循环次数的活性衰减变化。(b) 分别在j₁₀和j₅₀₀下的相应过电位。(c) HDFe-N-C与商用RuO₂的本征活性比较。(d) 不同电催化剂在j₅₀₀下未经iR校正的计时电位曲线。(e) HDFe-N-C和NiFe LDH未经iR补偿的AEMWE极化曲线。(f) 基于HDFe-N-C和NiFe LDH阳极的AEMWE在5Acm⁻²顺序电流密度下的计时电位曲线。图(f)中的插图展示了AEMWE电池结构。(g) HDFe-N-C NSs与近期报道的其他用于AEMWE的高性能OER基准催化剂的稳定性比较。
总之,该研究已成功证明,Fe负载量控制着Fe-N-C单原子催化剂表面*OH覆盖度,从而允许OER路径从AEM到OPM的可控转变。以OPM为主导的HDFe-N-C电催化剂在OER活性和稳定性方面均表现出显著优势:在500mAcm⁻²的电流密度下仅需288mV的过电位,并保持了长达2000小时的出色耐久性。当用作AEMWE中的阳极催化剂时,该系统展现出卓越的性能,在2.15V的电压下实现了5Acm⁻²的高电流密度,并可持续稳定运行超过500小时。此外,在动态启-停条件下,于0至5Acm⁻²范围内进行了35000次间歇电解循环,电压衰减率低至2.74mV kth⁻¹。该研究不仅为开发高性能、非贵金属OER催化剂提供了一个框架,也为单原子催化剂的理性设计和反应路径工程提供了新视角。这种创新的调控策略有望推动单原子催化剂在能源转换应用中的持续优化和性能提升。