郑州轻工业大学刘双良/南京大学朱少林:通过硼氮杂萘烯烃的不对称远程氢官能团化模块化构建手性萘生物电子等排体
- 2026-06-29 11:03:10

导语
近日,郑州轻工业大学刘双良联合南京大学朱少林课题组发展了一种通过硼氮杂萘烯烃的不对称原位及远程氢官能团化,来合成α-手性硼氮杂萘的通用策略。该方法基于合理设计的多功能PyrOx配体,可高效促进链行走与不对称偶联。DFT计算和机理实验表明,氧化加成是对映选择性的决定步骤。多种衍生化实验进一步验证了该方法的实用性。相关研究成果在线发表于CCS Chemistry(DOI: 10.31635/ccschem.026.202607942)。
背景及出发点
生物电子等排替代是药物发现与材料科学中的一项基本策略。鉴于萘骨架在药物分子中广泛存在,研究者持续探索其结构保留与性质优化的生物电子等排体。
作为一类芳香族硼氮杂环化合物,2,1-硼氮杂萘兼具可调得电子性质和氢键供体能力(图1A,上),有望改善靶标亲和力、生物活性及药代动力学特性。Rombouts等将普萘洛尔中的萘环替换为硼氮杂萘环,所得类似物在保持药效与 ADME-Tox特性的同时,展现出更优的生物利用度和血脑屏障穿透性(图1A,下)。该工作激发了官能团化硼氮杂萘的研究热潮,然而,当前高效、模块化合成手性硼氮杂萘的方法仍极为匮乏,严重限制了其后续应用探索(图1B)。
丰产金属催化的烯烃选择性氢官能团化反应取得的巨大进展,为手性2,1-硼氮杂萘的高效构建提供了潜在策略。在Song等报道的CuH催化不对称硼氢化反应的基础上(图1C,左),鉴于NiH催化可在烯烃原位和远程位点实现多种官能团的引入,作者拟开发一条合成手性2,1-硼氮杂萘的互补路线(图1C,右)。借助能够高效促进链行走和不对称偶联的多功能手性PyrOx配体,作者建立了一种温和的NiH催化体系,能够以易得的B-烯基-2,1-硼氮杂萘底物和溴代芳烃为原料,实现目标转化(图1D)。
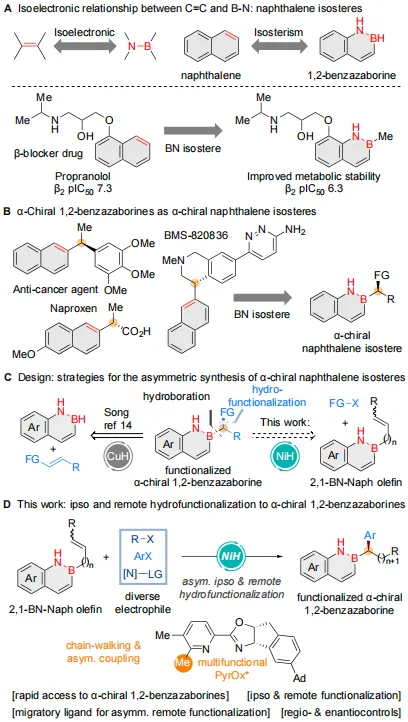
图1. 研究背景及思路(图源:CCS Chem.)
图文解析
反应优化
作者以反式-B-丙烯基-2,1-硼氮杂萘(1a)与4-溴苯甲酸甲酯(2a)为模板底物,对原位对映选择性氢芳基化反应进行了条件优化(图2)。通过对镍源、配体、硅烷、碱及溶剂的系统筛选,成功以良好收率、单一区域选择性和高对应选择性得到目标产物——α-芳基烷基-2,1-硼氮杂萘3a(entry 1)。当以NiI₂为催化剂时,收率和对映选择性均降低(entry 2)。PyrOx配体的选择尤为关键;该配体家族的其它成员(L2*−L4*)效果均不理想(entries 3−5)。噁唑啉环上的取代基(entries 3, 4)以及吡啶环C6位的取代基(entry 5)均对对映选择性有显著影响。进一步筛选表明,二甲氧基甲基硅烷作为氢源效果不佳(entry 6),CsF作碱时亦无改善(entry 7)。在单一溶剂中进行反应时,收率下降,但对映选择性基本不变(entries 8, 9)。升温至25 °C,收率略有提高,但对映选择性有所降低(entry 10)。活性更高的芳基碘化物虽可作为偶联试剂,但收率显著下降(entry 11)。烯丙基取代的2,1-硼氮杂萘(1a′)同样适用,能够以相当的收率和对映选择性得到迁移产物(entry 12)。克级规模实验效果相近,证实了该方法的实用性(entry 13)。
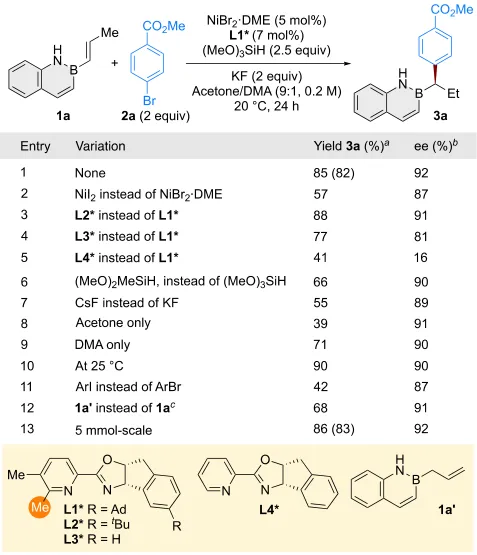
图2. 条件筛选(图源:CCS Chem.)
底物拓展
在优化反应条件下,作者首先考察了原位氢芳基化反应的底物适用范围(图3A)。多种芳基(3a−3r)和杂芳基(3s−3u)溴化物均可有效偶联,无论带有吸电子基团(3a−3l)还是供电子基团(3n−3r),均能顺利参与反应。该体系兼容多种官能团,包括酯基(3a, 3b, 3q)、酰胺基(3d, 3r)、三氟甲基(3h)、硅烷基(3l)、醚基(3n, 3o)和硫醚基(3p)。值得注意的是,易于被还原的酮(3e, 3f)和醛(3g)均保持完好。而具有潜在反应活性的硼酸酯(3c)、芳基氯(3i)、芳基对甲苯磺酸酯(3j)和芳基三氟甲磺酸酯(3k)也均完整保留,为后续衍生化提供了可能。该方法的实用性进一步体现在对结构复杂的生物活性天然产物及药物分子的后期官能化修饰中,如薄荷醇(3v)、吉非罗齐(3w)和氯贝特(3x)。同时,该反应对含多种取代基的2,1-硼氮杂萘底物均展现出优异的普适性(图3B),且稳定得到高对映选择性,包括供电子基团(4b−4f)和吸电子基团(4g−4k)。β位无取代的乙烯基底物(4l)同样有效。此外,β位带有不同取代基的烯基2,1-硼氮杂萘(4m−4o)也可顺利发生不对称氢芳基化。
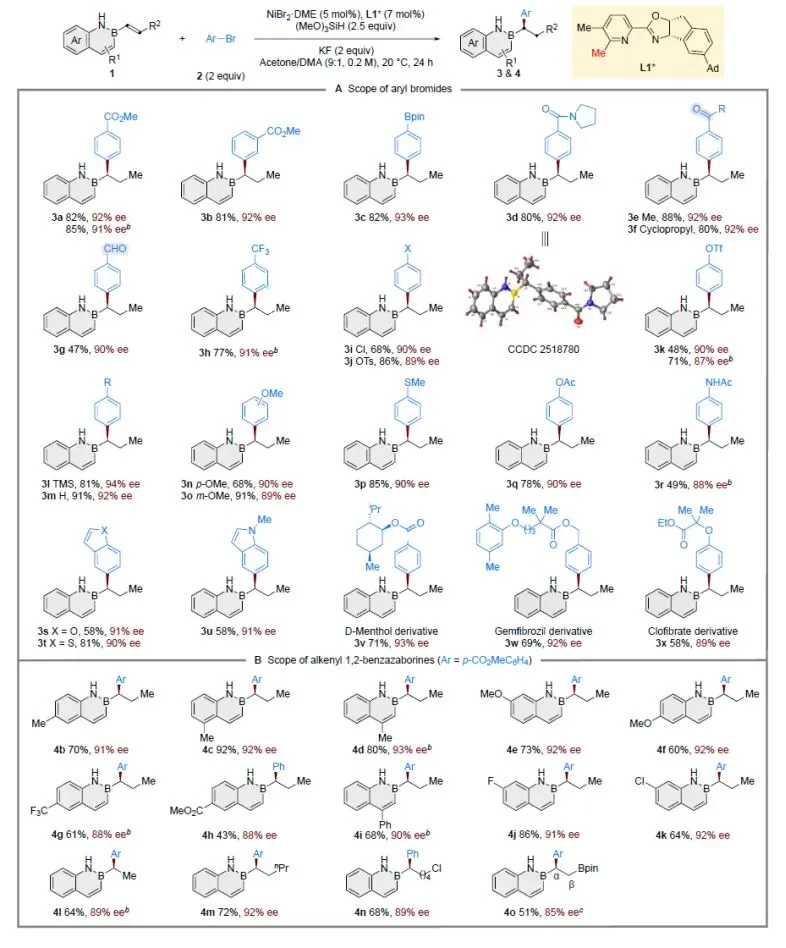
图3. 原位反应底物拓展(图源:CCS Chem.)
随后,作者探索了更具挑战性的含远程烯基的2,1-硼氮杂萘的不对称迁移氢芳基化反应(图4A)。在温和条件下,该反应专一性地发生于α-碳上,以良好收率和优异对映选择性得到产物。芳基亲电试剂方面,带有吸电子(7a−7f)或供电子基团(7g−7i)的芳基溴化物,以及杂芳基溴化物(7j, 7k),均能良好兼容。此外,多种硼氮杂萘环上带有不同取代基的惰性末端烯烃(8b−8l)和内烯烃(8m, 8n)均可作为有效底物参与反应(图4B)。
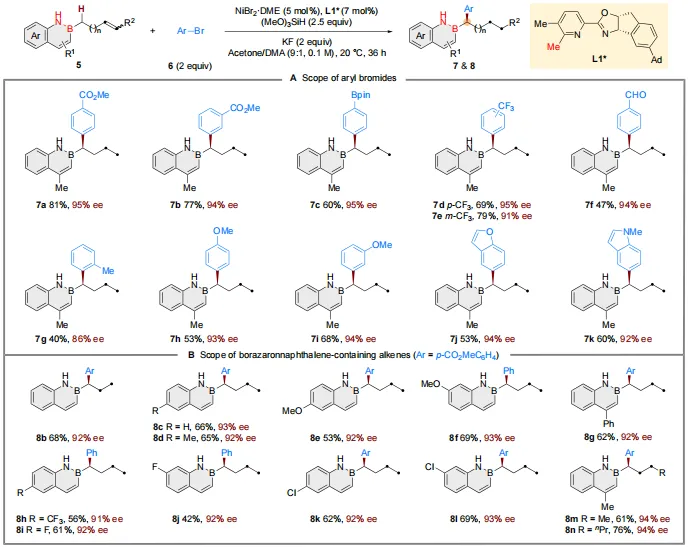
图4. 迁移反应底物拓展(图源:CCS Chem.)
反应应用
该方法的实用性通过制备具有生物活性的硼氮杂萘等排体3y和3z得以体现——二者分别为抗癌药物和维甲酸受体激动剂的活性类似物,充分展示了其在药物发现中的应用潜力(图5A)。此外,母体硼氮杂萘骨架还进行多种衍生化转化(图5B)。例如,3a经Ir催化C8−H硼化得到化合物9;经C3-碘代和钯催化交叉偶联得C3-芳基化产物10。化合物4n经分子内SN2环化可得到三环产物11,且对映选择性不受影响。为探究偶联位点的选择性,作者使用多种β-取代的硼氮杂萘底物进行了竞争实验(图5C)。结果表明,芳基化优先发生在与硼氮杂萘结构单元直接相连的α-碳上,而非靠近硼酸酯基团的α-碳(4o);且苄位优先于与硼氮杂萘相邻的α-碳(4p)。
机理研究
为明确氢镍化是否为对映选择性决定步骤,作者以Ph₂SiD₂为氢源进行了同位素标记实验(图5D)。若NiD物种对烯烃的迁移插入为决速步,则氘代产物3a-D应表现出高非对映选择性。但实验仅观察到约1:1的非对映异构体混合物,从而排除了氢镍化作为决速步的可能性。
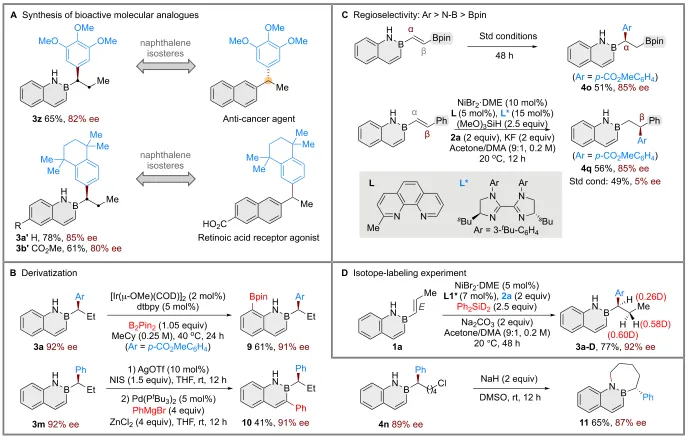
图5. 合成应用、区域竞争反应及氘代实验(图源:CCS Chem.)
密度泛函理论(DFT)计算表明,氧化加成是对映选择性决定步骤(图6)。手性烷基镍(I)中间体²Int1-R与芳基溴化物经选择性氧化加成生成手性烷基-Ni(III)-芳基中间体²Int3-R。该过程经由过渡态²TS2-R的能垒为8.1 kcal/mol,较经由²TS2-S的竞争路径低2.2 kcal/mol。过渡态的结构分析显示:²TS2-R中氢原子靠近配体上的叔丁基;而²TS2-S中丙基与叔丁基之间的位阻排斥使得该路径在能量上不利。未反应的²Int1-S可通过可逆的β-氢消除/再插入过程与²Int1-R快速互变。一旦²Int3-R不可逆生成,后续经²TS4-R的还原消除能垒仅为4.9 kcal/mol,即可得到最终产物。综上,氧化加成的选择性决定了整个转化路径的对映选择性。
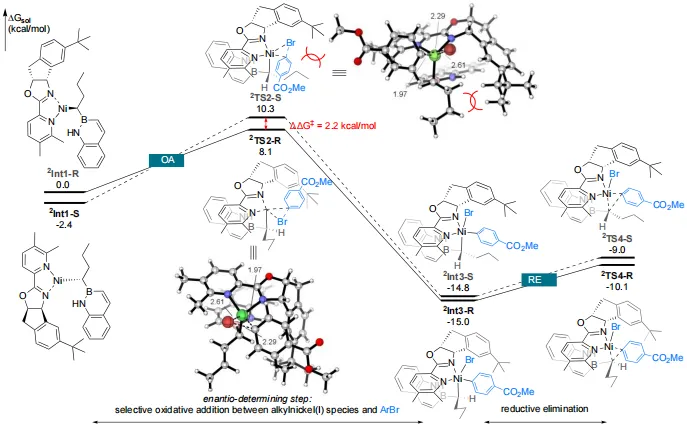
图6. DFT计算(图源:CCS Chem.)
总结
总之,作者利用NiH/PyrOx催化体系实现了烯烃原位及远程对映选择性氢芳基化,可在温和条件下高效合成α-手性2,1-硼氮杂萘,兼具优异的区域和立体选择性。该模块化策略为手性萘等排体的构建提供了简洁途径,在药物发现和材料化学中具有广阔应用前景。
该成果近期以“Modular Access to Chiral Naphthalene Isosteres via Asymmetric Remote Hydrofunctionalization of Borazaronnaphthalene Alkenes”为题发表于CCS Chemistry(DOI: 10.31635/ccschem.026.202607942)。郑州轻工业大学刘双良和南京大学在读博士生王嘉利为共同第一作者,通讯作者为南京大学朱少林/王优。

| 点击即可阅读合集 | ||
| 催化化学 | ||
| 分析化学 | ||
| 生物化学 |


