郑州大学蓝宇教授/张文静副教授/李世俊副教授/牛林彬副研究员Nature Communications: 从高能可见光催化到锰低能光氧化还原催化的演变
第一作者:杨威、宋雅雯、于雪晗
通讯作者:蓝宇教授、张文静副教授、李世俊副教授、牛林彬副研究员
通讯单位:郑州大学化学学院
原文链接:10.1038/s41467-026-68837-y
降低光催化反应中的能量输入是极具价值却充满挑战的目标,因为改变光催化剂的光吸收或催化特性通常需要繁琐的制备过程,导致研发周期漫长。本研究通过将简单的锰盐与廉价的具有配位能力的化学物质进行原位组装,实现了锰基低能光氧化还原催化,省去了复杂的预先制备步骤。将Mn(acac)2、2,2'-联吡啶-6,6'-二胺与TMSN3在反应体系中原位组装后,形成了可见光吸收体系。该体系在蓝光照射下可产生叠氮自由基,进而能以水为氢源驱动非活化烯烃的反马氏加氢叠氮化反应。基于此组装策略,进一步将Mn(acac)3与TMSN3在乙腈/六氟异丙醇混合溶剂中组合,构建出光吸收范围扩展至850纳米的催化体系;利用这一特性,成功实现了烯烃的一步法羟基叠氮化反应。这些发现为开发基于3d金属原位组装的低能光化学体系开辟了新途径。
传统光催化反应高度依赖高能光(λ<500nm),容易造成能源浪费,还可能导致反应物过度官能化、官能团不兼容等问题。同时,高能光穿透深度差、生物相容性低的缺陷,严重限制了其在放大合成与生物医学领域的应用。
3d金属储量丰富、毒性低且催化活性多样,将外源光能与3d金属催化结合,已成为丰富有机合成策略的重要方向。但当前3d金属基光氧化还原催化仍严重依赖高能光,而低能量光(λ>595nm)具备温和性与深组织穿透性,在有机合成、生物医学等领域极具应用潜力,开发3d金属基低能量光催化体系成为关键需求。
锰作为3d金属的重要成员,其光催化研究尚处于起步阶段。现有研究多聚焦于CO配位的Mn(0)配合物(如Mn2(CO)10)介导的碳中心自由基生成,而Mn(II)或Mn(III)基光催化体系生成氮中心自由基的研究仍属空白。氮中心自由基在有机合成中地位重要,因此开发可调控光能吸收、基于易得原料的Mn(II/III)基光催化平台,具有重要的科学价值。
1. 核心科学问题
如何突破传统光催化剂制备繁琐、依赖高能光的局限,开发无需复杂预制备、可直接利用低能量光的锰基光催化体系,实现高效、高选择性的有机转化反应。
2. 创新策略设计
提出原位组装策略,通过简单锰盐与廉价的具有配位能力的化学品的原位组合,调控配体、阴离子或金属氧化态,直接构建光吸收与催化活性中心,无需单独制备复杂光催化剂,快速实现从高能光催化到低能量光催化的转化。
3. 具体研究目标
基于原位组装策略,构建Mn(II)基高能可见光催化体系,实现未活化烯烃的反马氏氢叠氮化反应。优化组装体系,拓展光吸收范围至近红外区(850 nm),开发Mn(III)基低能量光催化平台,一步实现烯烃的羟基叠氮化反应。
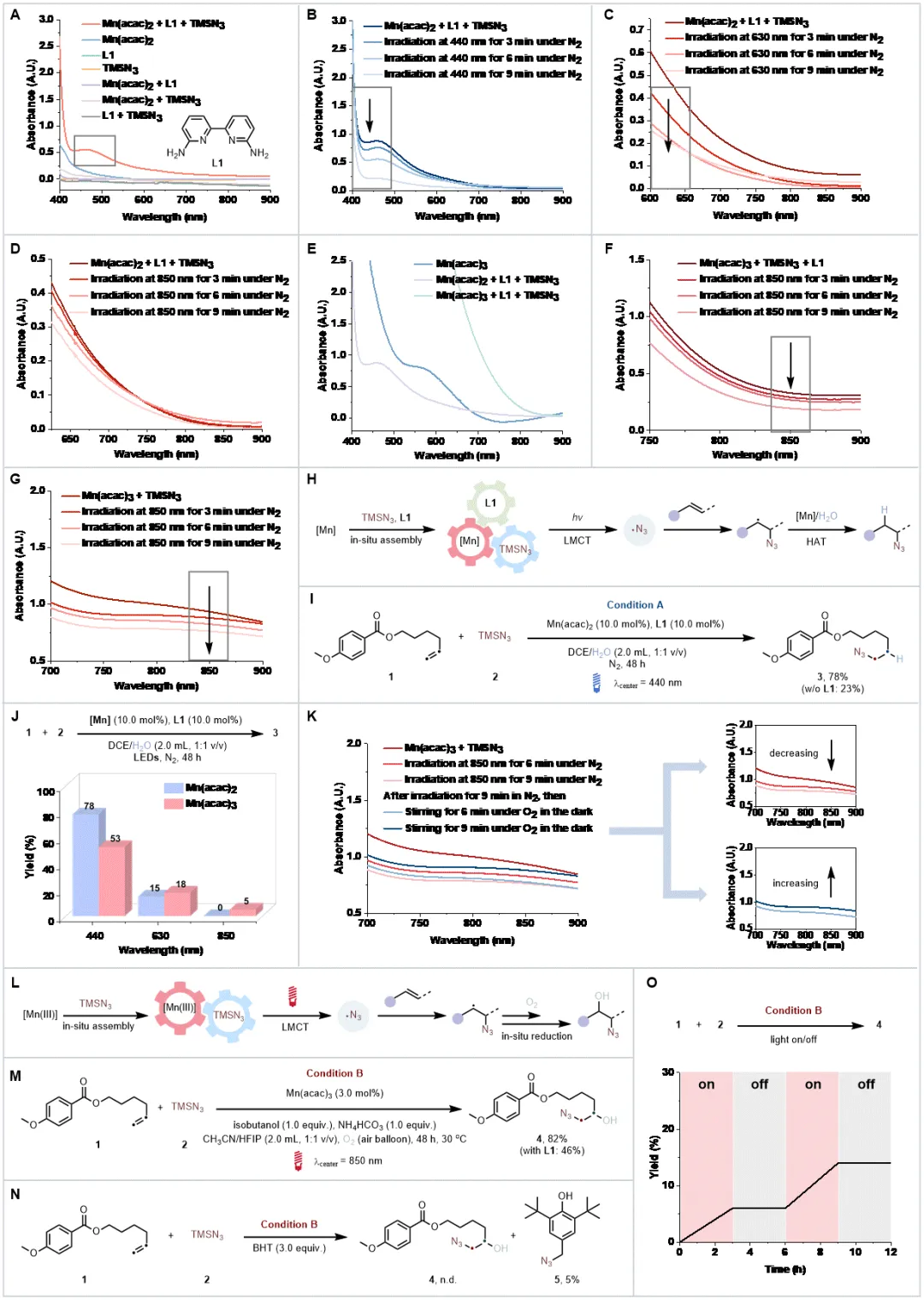
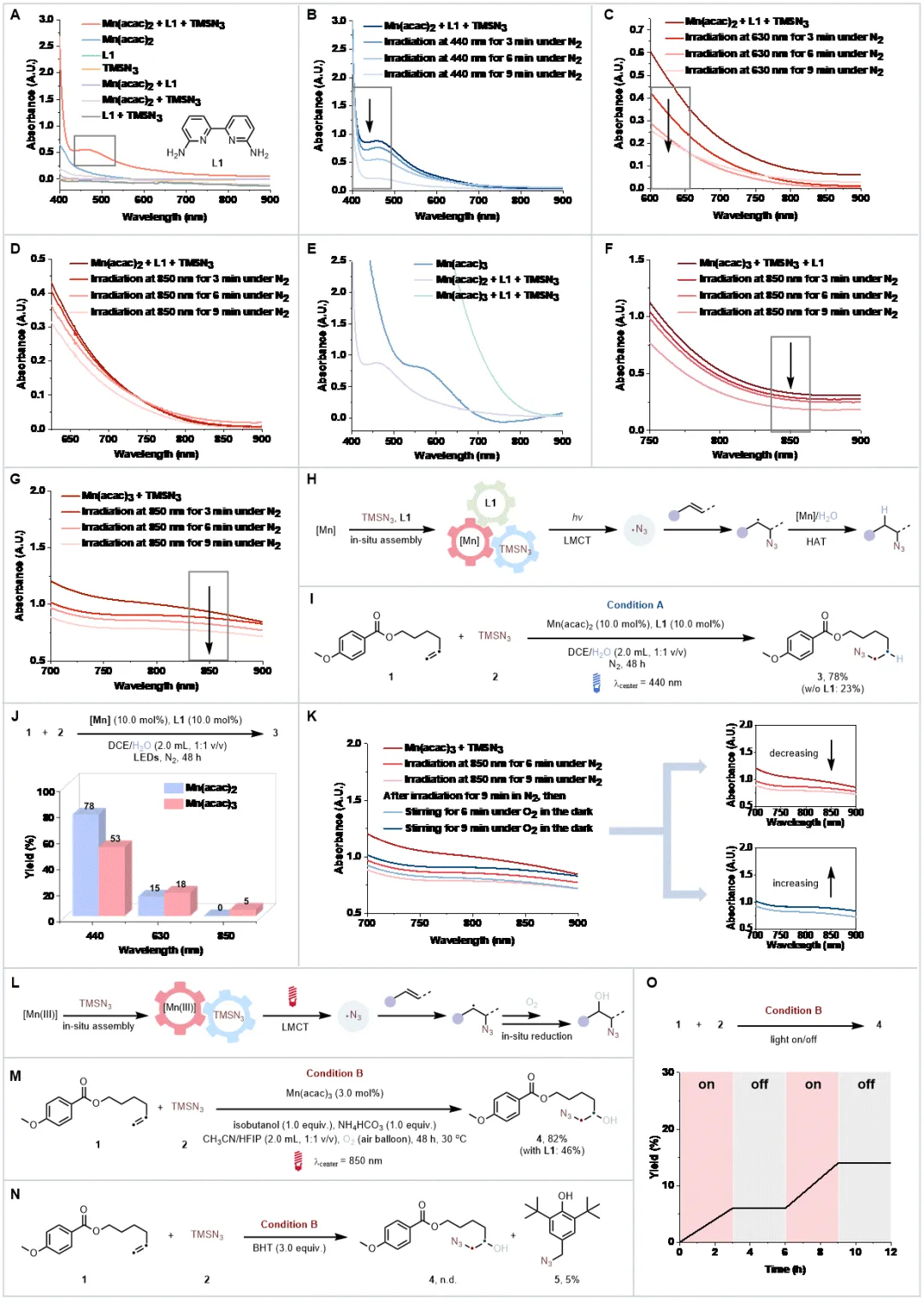
Mn(acac)₂与配体、TMSN₃原位组装形成的体系在 475 nm 处出现强吸收峰,且吸收尾端延伸至低能量区域,在440nm的蓝光照射下该吸收峰快速衰减,而在630nm和850 nm低能量光照射下,该体系虽能被消耗但效率显著降低,原因可能是其在700-850 nm区域吸光较弱;将锰价态提升至+3(采用Mn(acac)3)后,体系在700-850 nm低能量区的吸光强度显著增强,且在850 nm近红外光照射下吸光度会明显下降。进一步的研究发现,配体对近红外光吸收并非必需;以反马氏加氢叠氮化为模型反应,Mn(II)体系在440 nm蓝光下实现78%的产率,H2O经D₂O替代实验证实为氢源。而Mn(III)体系在该反应中因无外部氧化剂时难以再生导致产率极低;引入绿色氧化剂O2后,Mn(III)体系可顺利再生,结合异丁醇作为还原剂,在850 nm低能量光下一步实现烯烃的羟基叠氮化,目标产物产率达82%,自由基捕获实验(BHT)证实叠氮自由基是关键中间体,开关实验则排除了自由基链式反应路径。
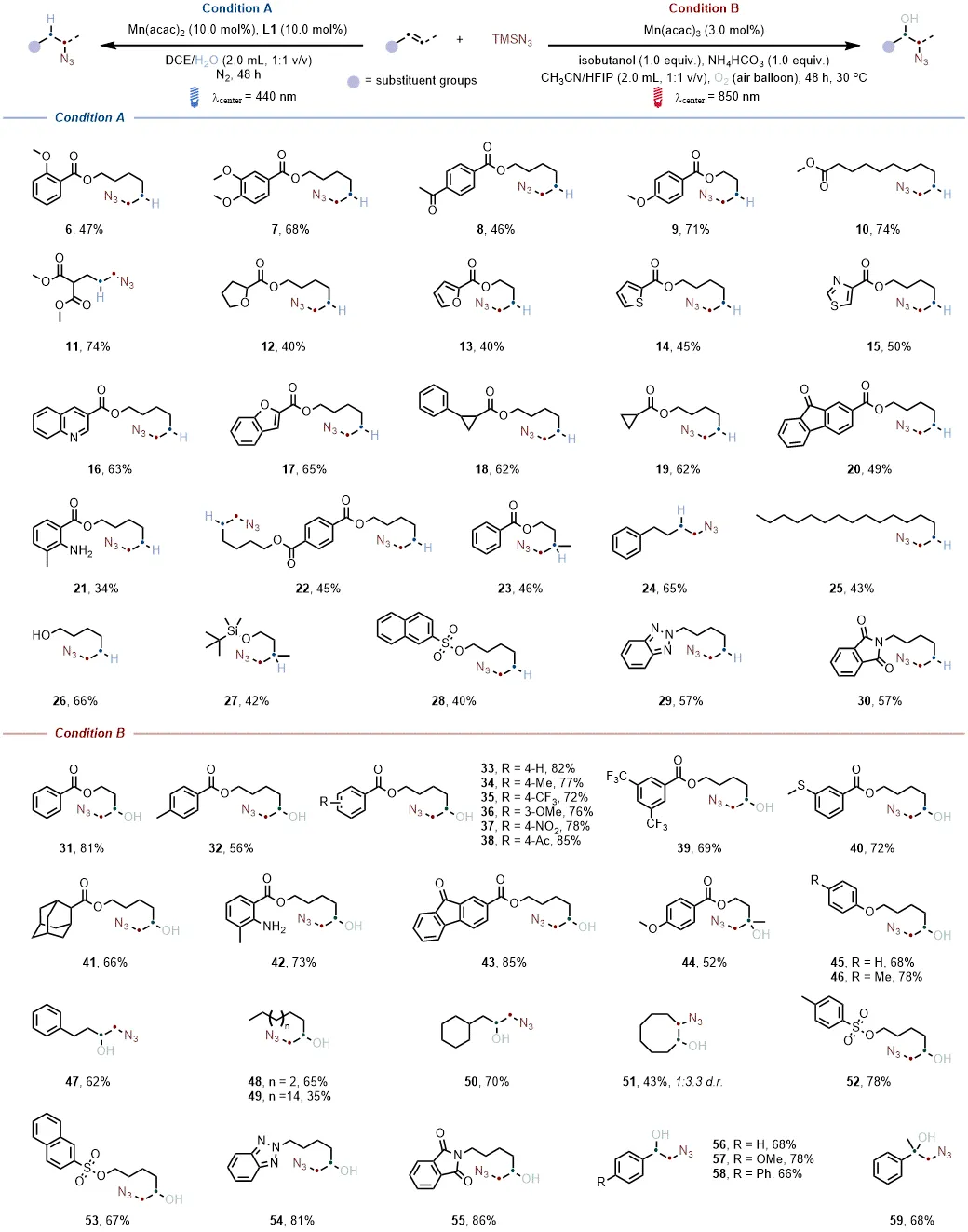
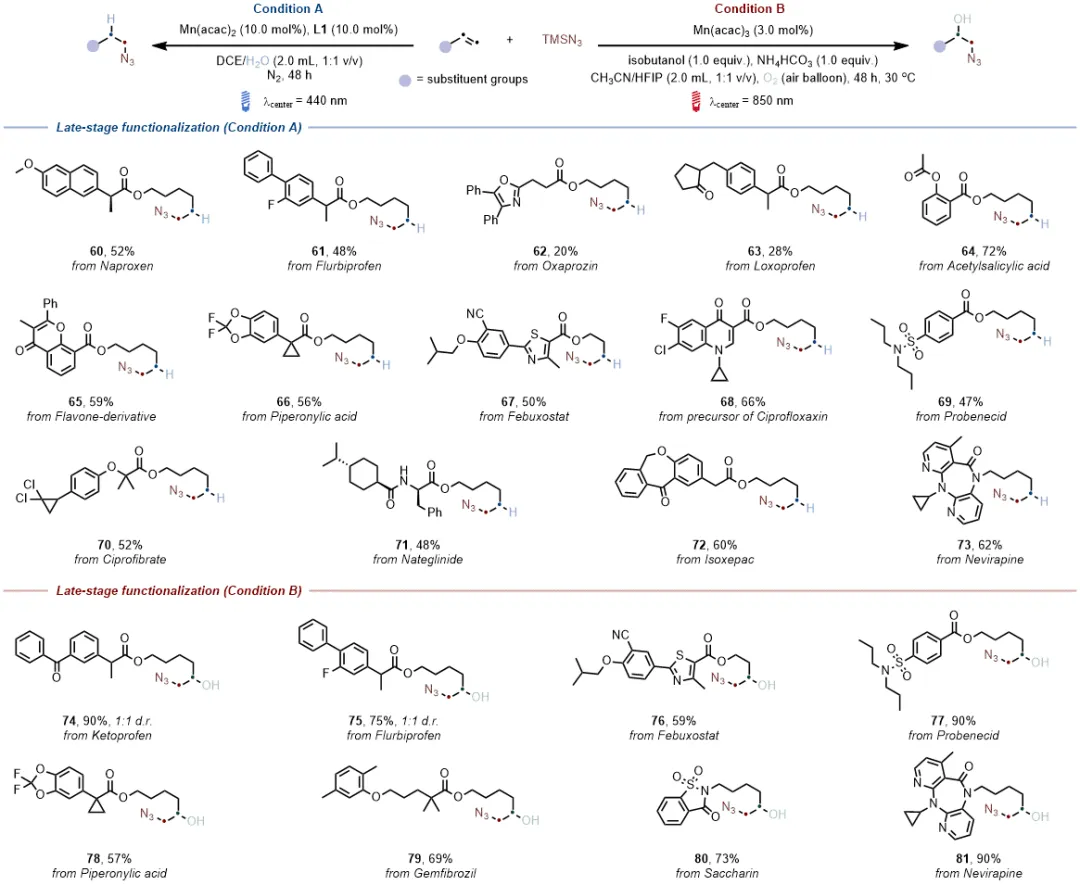
底物拓展发现多种烯烃均能在对应催化条件下高效转化为附加值烷基叠氮化物,产率处于中等至良好水平,即便存在喹啉、硫醚、未保护胺、醇、噻唑、三唑等可能影响原位组装的官能团。含CF₃基团、杂环结构及环丙基的烯烃,催化反应仍能顺利进行,展现出优异的官能团耐受性,其中CF₃基团与杂环的引入还提升了产物在生物医药领域的应用潜力。
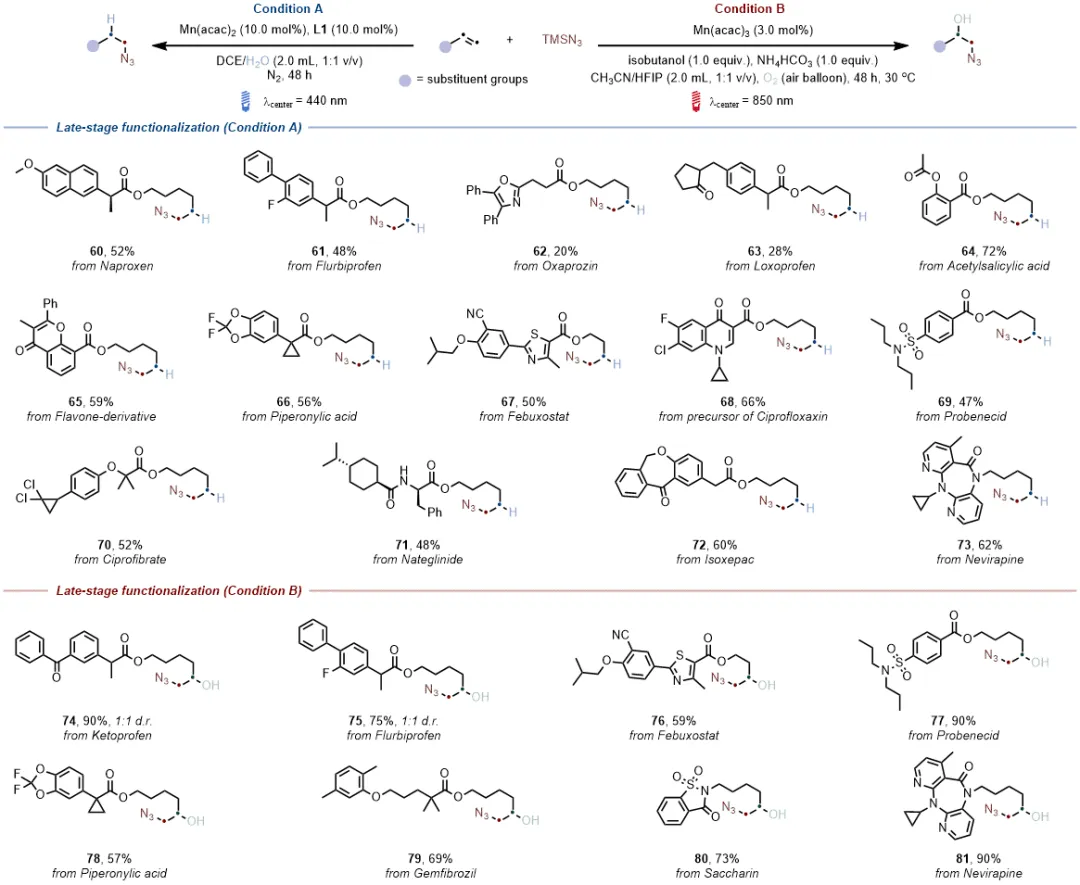
两种原位组装体系均能成功对多种含复杂分子骨架的药物烯烃进行修饰,生成复杂烷基叠氮化物,产率介于20%-90%之间,且低能量光催化体系生成的产物同时含有叠氮与羟基官能团,为生物医药研究提供了新的分子靶点,进一步验证了体系在药物研发领域的实用价值。
加氢叠氮化与羟基叠氮化物反应均能实现克级规模合成,且低能量光催化羟基叠氮化反应可直接利用太阳光驱动,凸显绿色环保优势;含蒽醌基团的烯烃因自身能吸收高能光,在高能光催化条件下无法发生碳碳双键的加氢叠氮化,而在630 nm或850 nm低能量光催化条件下可高效转化为β-叠氮醇,直观体现了低能量体系独特的官能团耐受性;催化性能对比实验表明,4CzIPN、Ir/Ru等传统贵金属光催化剂在该类反应中活性不佳。锰基体系的催化效果显著优于铁基体系;此外,加氢叠氮化与羟基叠氮化产物可进一步衍生为多种含氮有机化合物,极大拓展了合成应用场景,充分证明了该催化方案的实用价值。
本研究通过原位组装策略,成功实现了锰基光催化体系从高能可见光(440 nm)到低能量近红外光(850 nm)的演进。Mn(II)基高能光体系以H2O为氢源,高实现未活化烯烃反马氏加氢叠氮化反应;Mn (III)基低能量光体系通过O₂辅助再生,一步实现烯烃的羟基叠氮化反应。两大体系均具有优异的官能团耐受性、底物普适性,可规模化合成且能应用于药物分子后期官能化,性能优于传统贵金属光催化剂及铁基体系,为3d金属基低能量光化学的发展奠定了基础。
Wei Yang+, Yawen Song+, Xuehan Yu+, Meng Tian, Yu Lan*, Ya-Nan Wang, Xiaoyu Jiang, Shihan Liu, Wenjing Zhang*, Shi-Jun Li*, and Linbin Niu*. Evolution of manganese low-energy photoredox catalysis from high-energyvisible light photocatalysis.Nat. Commun.2026, doi: 10.1038/s41467-026-68837-y.