Advanced Science | 郑州大学发现活化血小板释放的HSP90α通过TLR4/MyD88/Beclin 1轴诱导自噬依赖性NETs形成加重脓毒症
- 2026-03-31 09:55:52
Metadata Card
文献题目:Activated Platelet–Released Heat Shock Protein 90α Triggers Autophagy-Dependent Neutrophil Extracellular Trap Formation and Amplifies Sepsis
发表期刊:Advanced Science
发表时间:26.2.24 Online
影响因子:14.1(中科院1区)
通讯作者/单位:Xiangzhan Zhu @ 郑州大学附属儿童医院
一句话总结:该研究发现脓毒症中血小板通过TLR4依赖途径从巨噬细胞继承并释放HSP90α,通过激活中性粒细胞TLR4/MyD88/Beclin 1自噬通路诱导NETs形成,进而加剧血栓炎症及器官损伤。
Graphical Abstract

Abstract
中文摘要:
血小板对脓毒症中血栓形成和凝血异常的发展至关重要,但它们促进这些病理过程的机制尚未完全了解。在这里,我们确定了血小板释放的热休克蛋白90 α(Hsp90 α)在驱动中性粒细胞细胞外陷阱(NET)形成和支持脓毒症期间血栓炎症中的关键作用。脓毒症患者血小板的蛋白质组学分析显示HSP90 α显著增加,我们将其追溯到源自巨核细胞的运输途径。当活化时,血小板将Hsp90 α转运至其质膜,并以游离形式和外泌体相关形式将其释放至细胞外空间。细胞外Hsp90 α作为与中性粒细胞上的toll样受体4(TLR4)结合的损伤相关分子模式。这种结合激活下游MyD88-Beclin 1信号通路,触发自噬并导致NET形成。用中和性单克隆抗体阻断细胞外Hsp90 α显著减少了体外和体内的NET形成,从而减少了脓毒症相关的血栓形成和炎症。这种血小板-HSP90 α-TLR4-自噬-NET途径不仅加深了我们对血小板诱导的免疫血栓形成的理解,而且为旨在减少脓毒症患者凝血问题和器官衰竭的治疗提供了潜在的靶点。
英文摘要:
Platelets are crucial to the development of thrombosis and coagulation abnormalities in sepsis, but the mechanisms by which they contribute to these pathological processes are not fully understood. Here, we identify a key role for platelet-released heat shock protein 90α (HSP90α) in driving neutrophil extracellular trap (NET) formation and supporting thromboinflammation during sepsis. Proteomic analysis of platelets from patients with sepsis showed a significant increase in HSP90α, which we traced back to trafficking pathways originating from megakaryocytes. When activated, platelets translocate HSP90α to their plasma membrane and release it into the extracellular space in both free and exosome-associated forms. Extracellular HSP90α acts as a damage-associated molecular pattern that binds to toll-like receptor 4 (TLR4) on neutrophils. This binding activates a downstream MyD88–Beclin 1 signaling pathway, triggering autophagy and leading to NET formation. Blocking extracellular HSP90α with a neutralizing monoclonal antibody significantly reduced NET formation both in vitro and in vivo, resulting in decreased sepsis-related thrombosis and inflammation. This platelet–HSP90α–TLR4–autophagy–NET pathway not only deepens our understanding of platelet-induced immunothrombosis but also suggests potential targets for therapies aimed at reducing coagulation problems and organ failure in septic patients.
Background & Problem
背景:败血症是一种因宿主对感染反应失调导致器官功能障碍的危重病症,常并发弥散性血管内凝血 (DIC) 等严重凝血障碍。血小板和中性粒细胞胞外诱捕网 (NETs) 是免疫血栓形成的共同参与者,两者在败血症中均显著激活并相互促进。
痛点:尽管已知血小板能通过释放vWF、PF4等介质促进NETs形成,但在败血症特定条件下,血小板启动或放大NETs形成的精确分子机制仍不明确。此外,传统的抗血小板药物(如阿司匹林)在败血症患者(尤其是伴有血小板减少症的患者)中存在极高的出血风险,亟需针对致病性因子而非血小板本身功能的精准疗法。
Methodology
策略:研究团队结合了临床样本蛋白质组学、单细胞测序数据挖掘、基因敲除小鼠模型以及药理学干预手段。
逻辑:
1.通过DIA-MS蛋白组学对比健康人与败血症患者血小板,锁定差异蛋白HSP90α。
2.利用盲肠结扎穿刺 (CLP) 和LPS诱导的败血症小鼠模型,验证HSP90α的来源(巨核细胞)及其在血栓形成中的作用。
3.通过免疫共沉淀(Co−IP)、透射电镜(TEM)和Confocal显微成像,解析胞外HSP90α诱导中性粒细胞自噬并形成NETs的具体信号通路。
4.最后,使用中和抗体1G6-D7进行干预,评估其在体内减轻血栓和炎症的治疗潜力。
Key Results & Interpretation
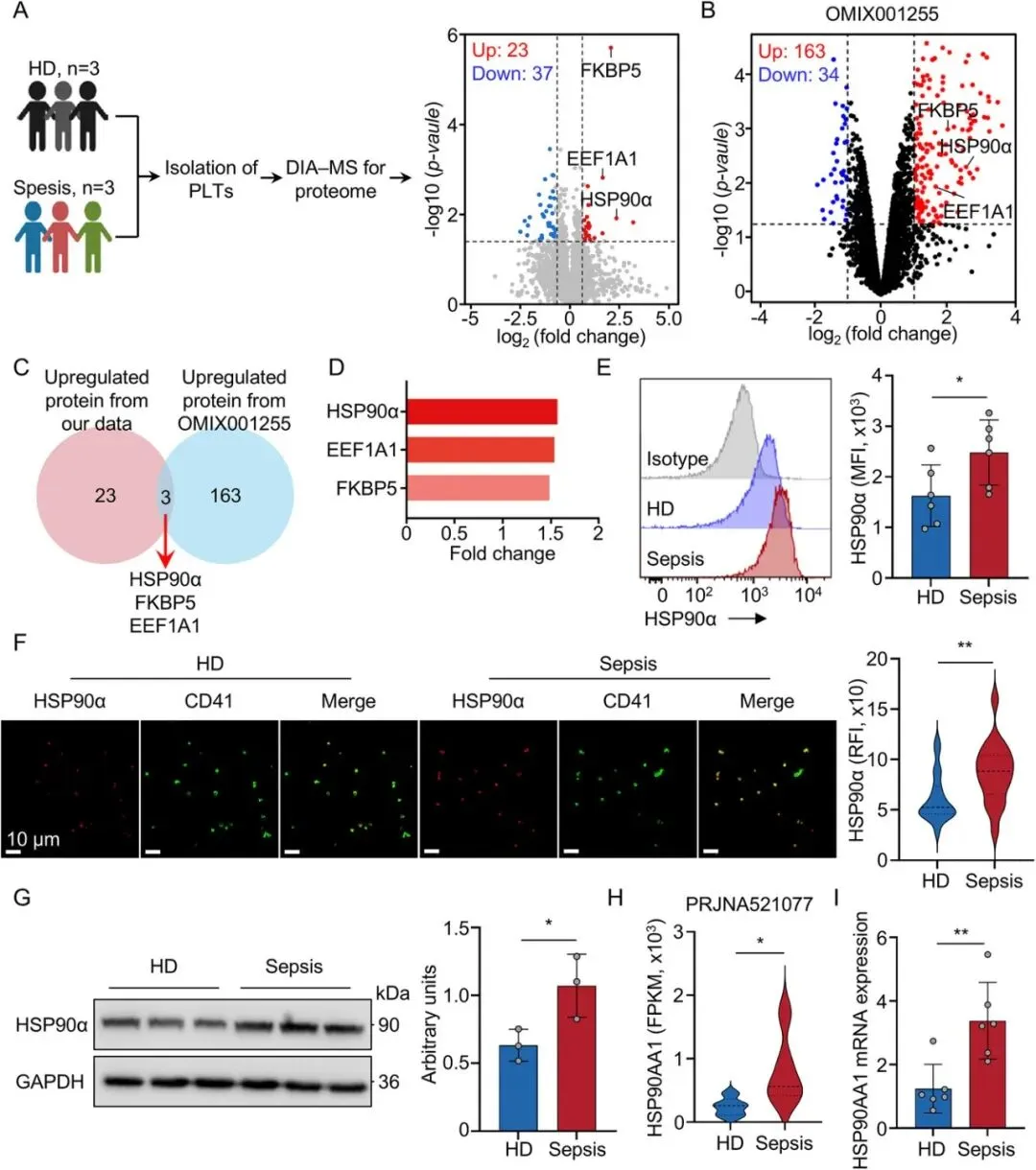
[A-B]:研究者对 3 例健康捐赠者(HD)和 3 例败血症患者的血小板进行了 DIA-MS 蛋白组学分析,在鉴定的 4627 个蛋白中发现 23 个显著上调。结合公开数据库 OMIX001255 的验证,确认了败血症状态下血小板蛋白谱的显著改变。
[C-D]:通过两个数据集的交集分析,锁定了三个共同上调蛋白:HSP90α、FKBP5 和 EEF1A1。其中,HSP90α 在败血症组中表现出最高的倍数变化。
[E-G]:利用流式细胞术 (MFI)、免疫荧光 (IF) 和 Western Blot (WB) 进行验证,证实败血症患者血小板内的 HSP90α 蛋白水平显著高于健康对照。
[H-I]:分析公开的 RNA-seq 数据 (PRJNA521077) 并结合 qRT-PCR 发现,血小板中编码 HSP90α 的基因 HSP90AA1 mRNA 同样显著升高。
小结:由于血小板无核且其 RNA 继承自巨核细胞 ,研究者进一步证实了败血症环境下巨核细胞中 HSP90α 的转录增强是血小板高表达的源头 。接下来的实验重点转向这些蛋白是如何从血小板中释放出来的。
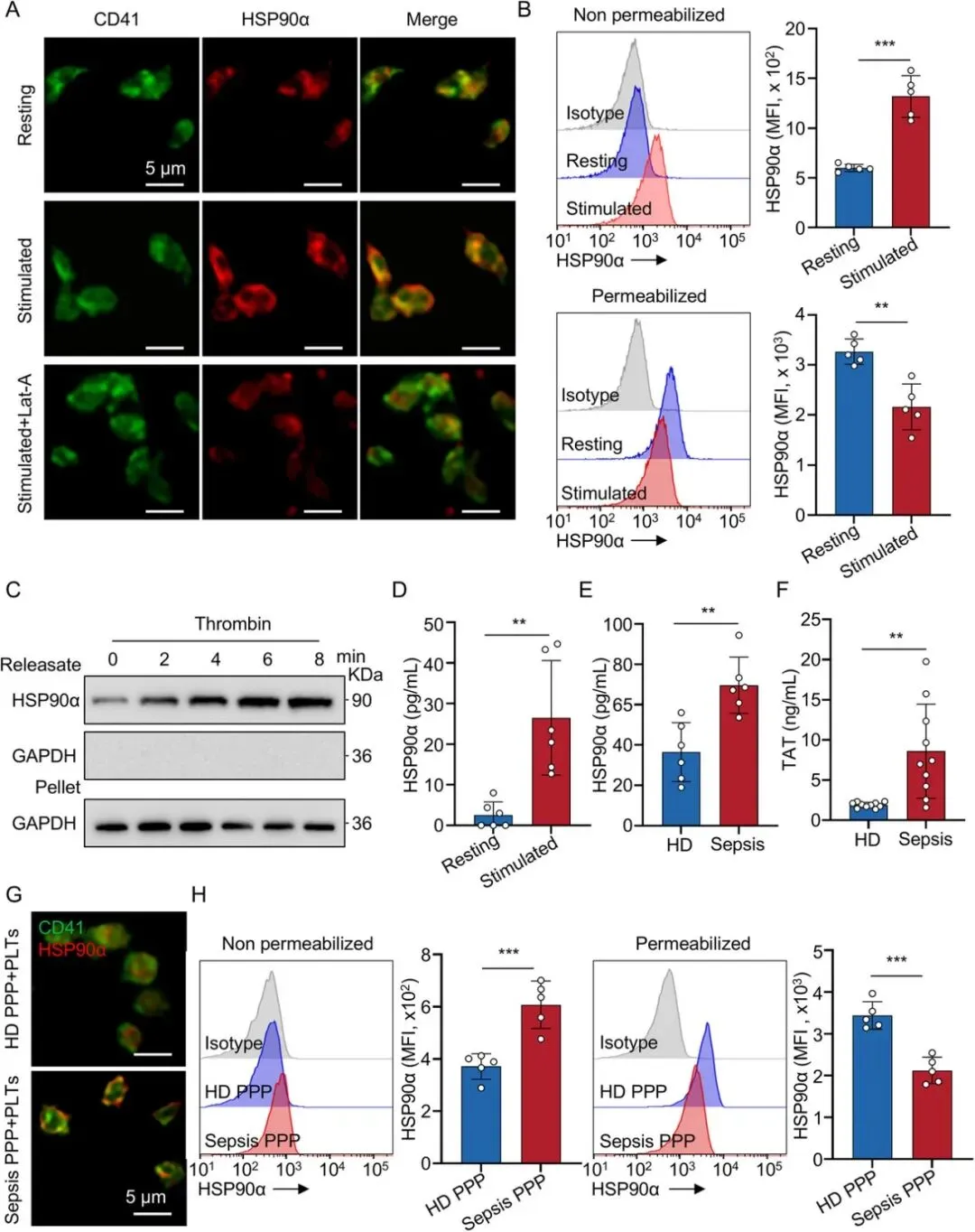
[A]:免疫荧光成像显示,静息血小板中的 HSP90α 弥散在胞质内。经凝血酶(Thrombin)刺激后,蛋白向质膜易位形成特征性的环状模式,且该过程依赖于肌动蛋白聚合。
[B]:流式细胞术对比透化与未透化的血小板,量化证实活化后胞内 HSP90α 含量减少,而膜表面表达量显著增加。
[C-D]:通过对血小板释放液(Releasate)的 WB 检测和上清液的 ELISA 分析,证实 HSP90α 在刺激后被释放到胞外空间。
[E-F]:对比发现败血症患者血小板释放 HSP90α 的水平显著高于健康组;同时,患者血浆中的 TAT 复合物(凝血酶活性标志物)水平显著升高,反映了体内强烈的促凝环境。
[G-H]:使用败血症患者的乏血小板血浆(PPP)直接处理健康血小板,诱导了 HSP90α 的膜易位和释放,证明循环中的可溶性促炎因子足以触发此过程。
小结:确立了 HSP90α 是活化诱导的分泌蛋白(eHSP90α)后 ,研究者开始探究这些释放到血浆中的蛋白是否具有致病功能,尤其是对血栓形成的影响。
发现点3:eHSP90α 介导的血栓形成高度依赖中性粒细胞的参与
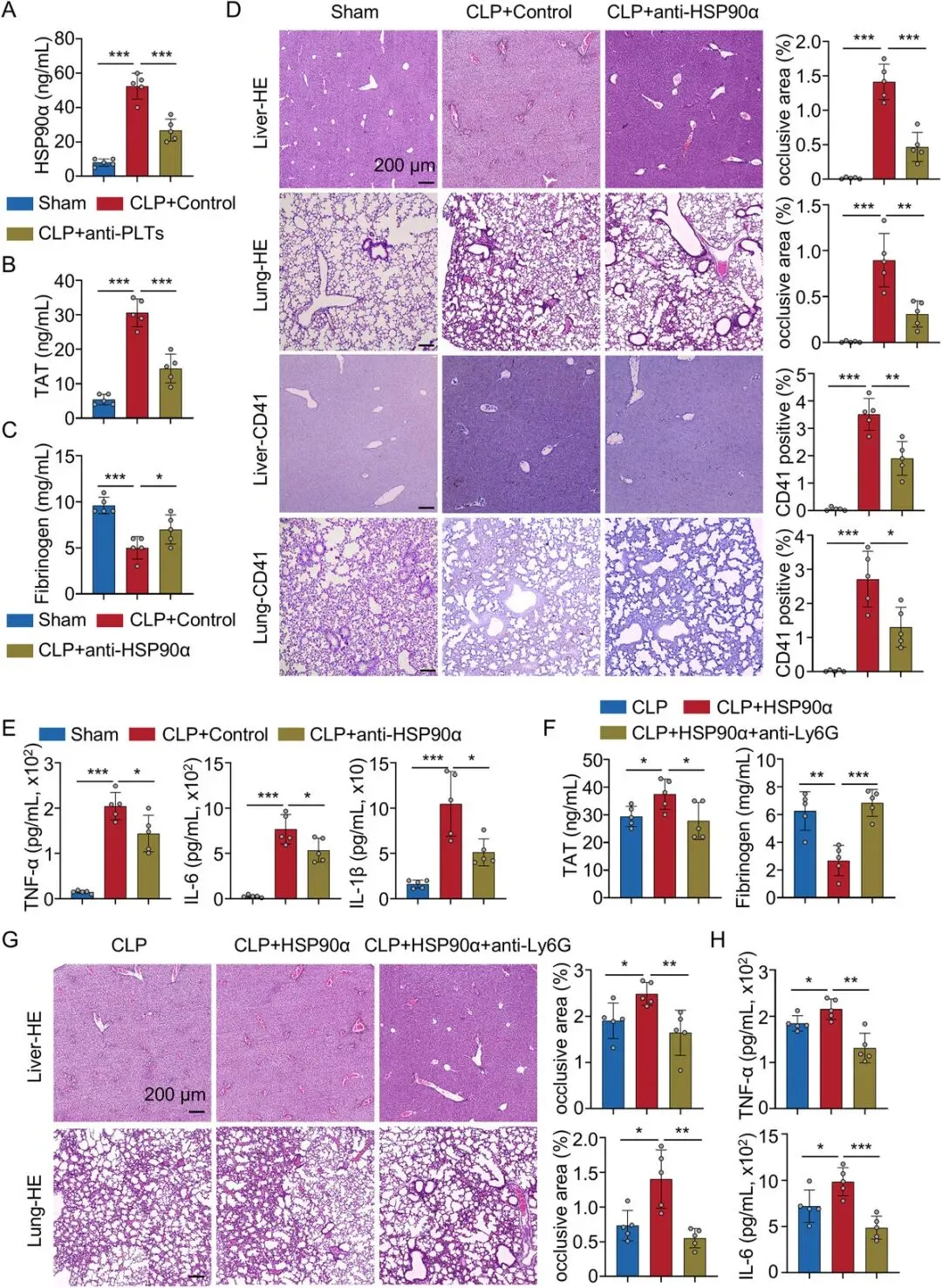
[A]:在 CLP 败血症小鼠中,清除血小板使血浆 eHSP90α 水平下降约 49.22%,证实血小板是循环 eHSP90α 的重要来源。
[B-C]:给予 1G6-D7(HSP90α 中和抗体) 干预后,败血症小鼠的 TAT 水平显著降低,纤维蛋白原水平得到恢复,表明凝血紊乱得到缓解。
[D]:肝、肺组织的 HE 染色和 CD41 免疫组化(IHC) 显示,中和抗体显著减少了微血管内的血栓形成和血小板聚集。
[E]:中和抗体同时显著降低了血浆中 TNF-α、IL-6 和 IL-1β 等促炎因子的水平。
[F-H]:为锁定效应细胞,研究者在补充重组 HSP90α 的同时使用了抗 Ly6G 抗体清除中性粒细胞。结果显示,缺乏中性粒细胞后,HSP90α 诱导凝血激活和器官血栓的能力被显著削弱,证明中性粒细胞是 eHSP90α 促栓作用的必需中介。
小结:数据将矛头指向了中性粒细胞。由于中性粒细胞在血栓中的主要贡献是形成 NETs,下一步实验聚焦于 HSP90α 是否直接诱导 NETs 产生。
发现点4:eHSP90α 剂量依赖性诱导 NETs 形成,且与病理严重程度正相关
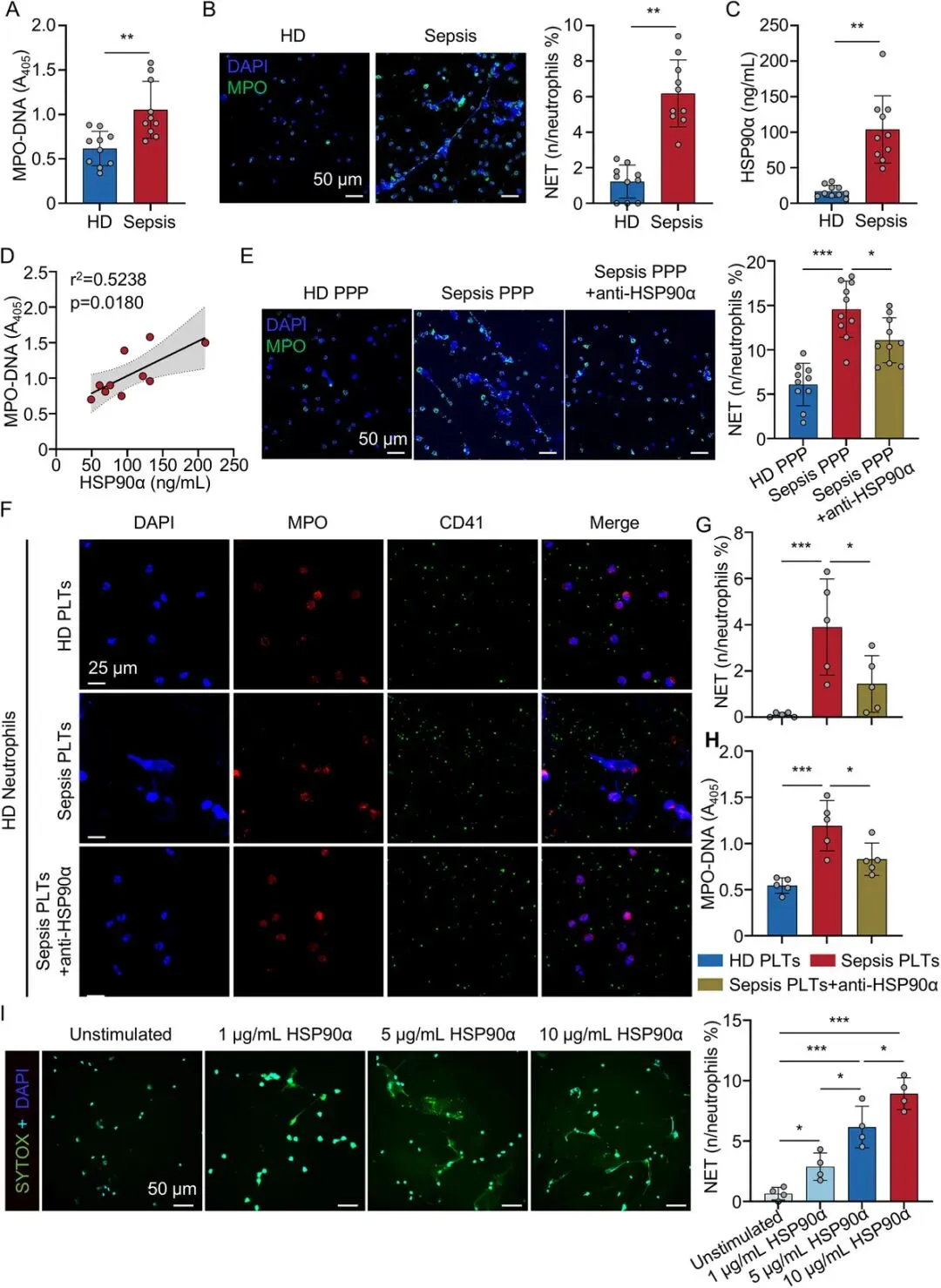
[A-C]:检测发现败血症患者血浆中 MPO-DNA 复合物(NETs 指标)和 HSP90α 水平均显著高于健康对照组。
[D]:相关性分析显示,患者血浆 HSP90α 浓度与 MPO-DNA 浓度呈显著正相关。
[E]:败血症 PPP 可诱导健康中性粒细胞产生 NETs,而加入 1G6-D7 抗体后,这一效应被显著抑制。
[F-H]:在血小板-中性粒细胞共培养体系中,败血症血小板诱导 NETs 的能力显著增强,且该过程可被 HSP90α 中和抗体阻断。
[I]:使用不同浓度的重组 HSP90α 直接刺激中性粒细胞,观察到 Sytox+ 荧光(DNA 释放)随浓度升高而增加,确证了其直接诱导能力。
小结:明确了“蛋白-细胞-效应”关系后 ,研究者进入分子机制层面,探索 NETs 形成的胞内核心通路——自噬。
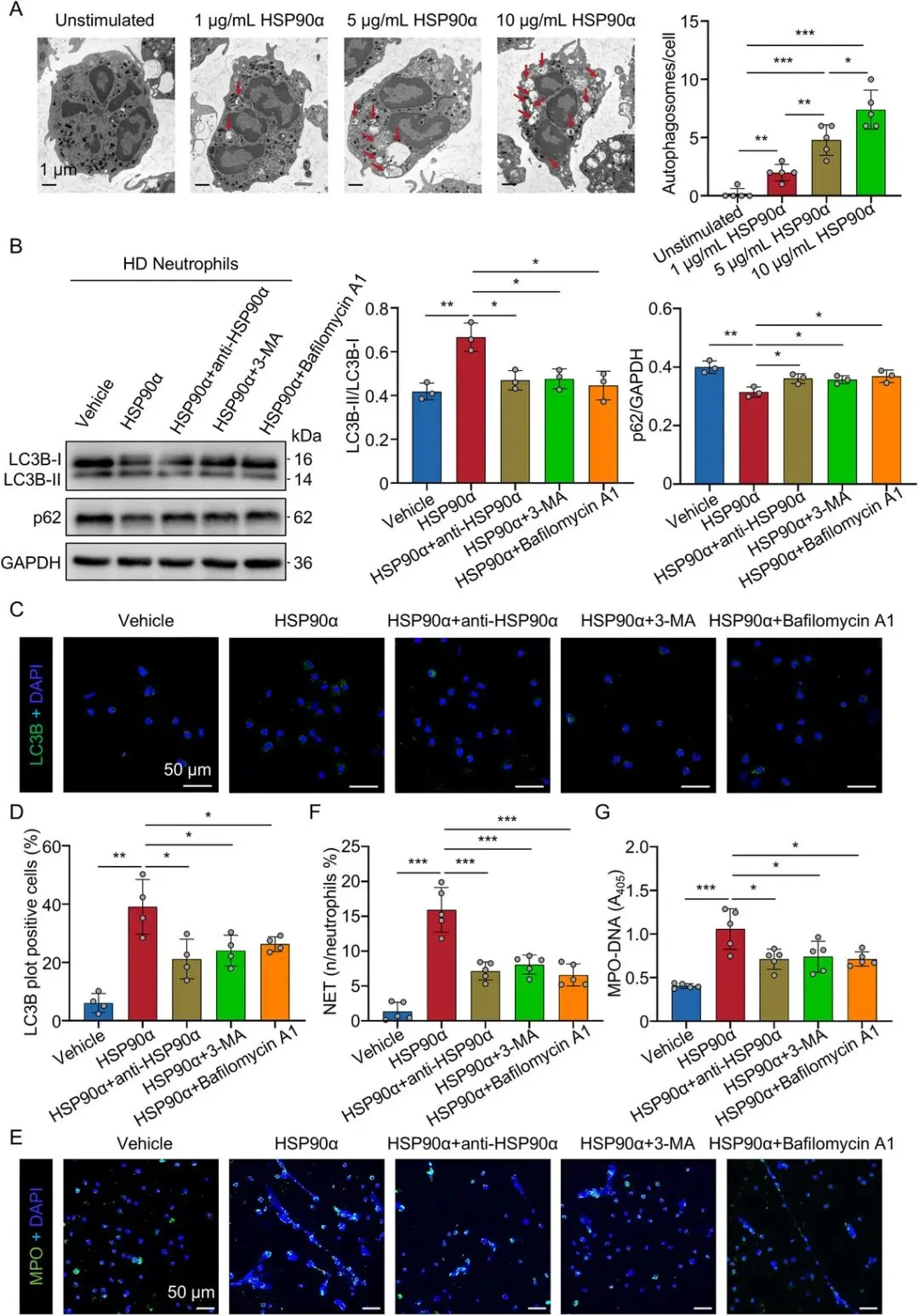
[A]:透射电镜(TEM) 观察到,经 HSP90α 刺激的中性粒细胞胞质内出现了大量自噬小体和自噬溶酶体。
[B]:WB 检测显示 LC3B-II/I 比值升高 且 p62 蛋白水平下降,证明了自噬流的整体激活。
[C-D]:免疫荧光统计显示 HSP90α 处理后 LC3B 阳性点显著增加,而加入自噬抑制剂 3-MA 或 Bafilomycin A1 可逆转此现象。
[E-G]:关键的功能抑制实验表明,阻断自噬能有效抑制 HSP90α 诱导的 MPO-DNA 释放和 NETs 结构形成。
小结:自噬是中间环节,那么胞外的 HSP90α 是如何跨越细胞膜启动这套机器的?最后一部分实验锁定了膜受体。
发现点6:eHSP90α 结合 TLR4 受体并经 MyD88/Beclin 1 轴触发通路
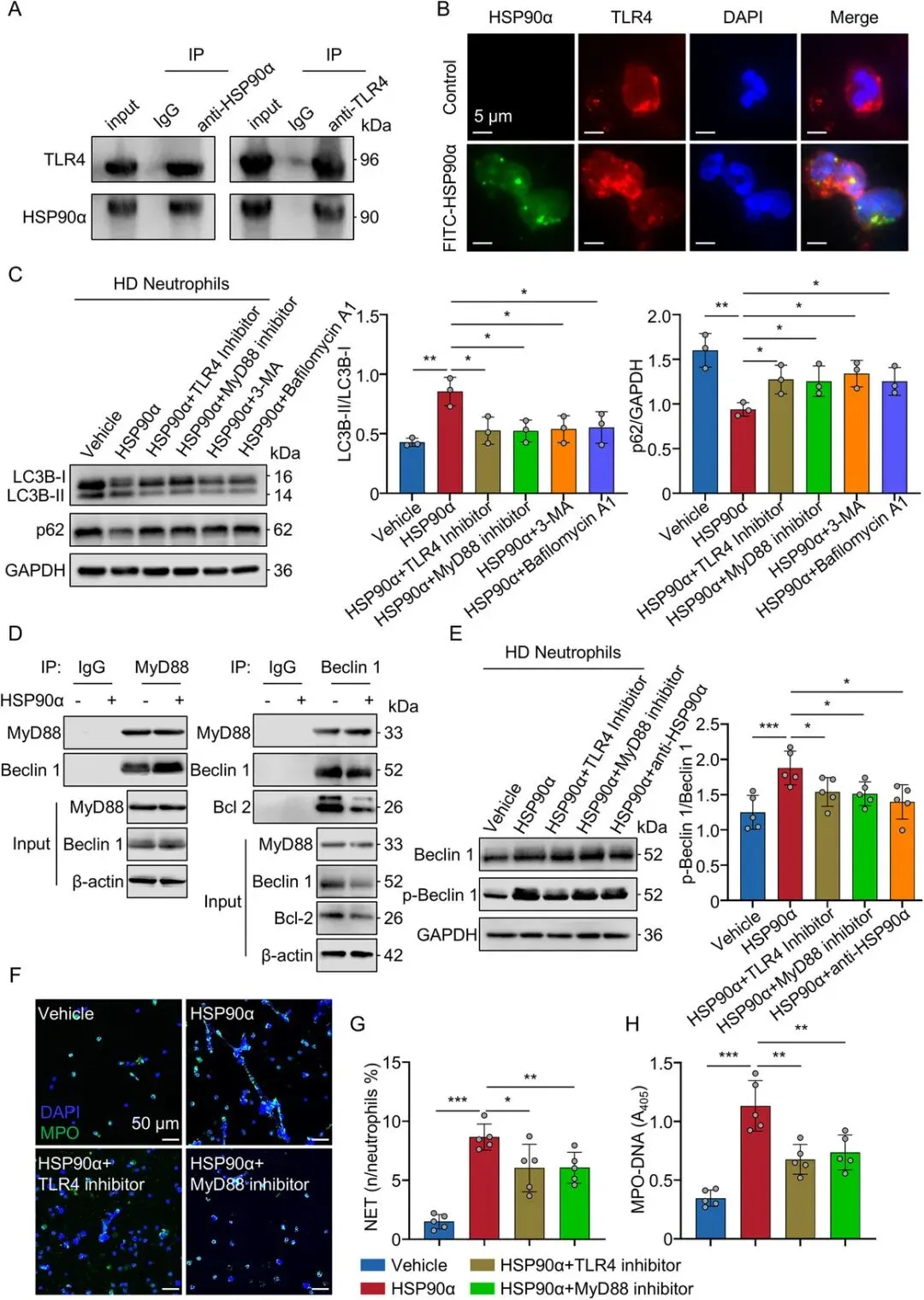
[A-B]:免疫共沉淀(Co-IP) 和荧光共定位实验证实,eHSP90α 能与中性粒细胞表面的 TLR4 受体胞外域直接结合。
[C]:使用 TLR4 抑制剂或 MyD88 抑制剂 处理后,HSP90α 诱导的自噬激活(LC3B-II 积累和 p62 恢复)被阻断。
[D]:Co-IP 发现刺激后 MyD88 与 Beclin 1 的相互作用增强,同时促进了 Beclin 1 与抑制因子 Bcl-2 的解离。
[E]:检测到 HSP90α 诱导了 Beclin 1 的磷酸化(p-Beclin 1),且这一过程受 TLR4 和 MyD88 的调控。
[F-H]:最终功能验证显示,药理学阻断 TLR4 或 MyD88 通路可使中性粒细胞对 HSP90α 诱导的 NETs 形成产生抵抗。
小结:败血症环境下活化血小板释放的 HSP90α,作为 DAMP 结合中性粒细胞表面的 TLR4,通过 MyD88/Beclin 1 信号轴激活自噬,最终诱发 NETs 形成并放大血栓炎症。
Innovation & Takeaway
✨ 创新点
1、揭示了“血小板-eHSP90α-中性粒细胞自噬-NETs”这一完整的败血症血栓炎症通路。
2、明确了败血症中血小板HSP90α的高表达源于其在巨核细胞阶段受TLR4信号驱动的转录增强。
3、筛选出的中和抗体1G6-D7在不干扰血小板基本功能的情况下显示出良好的抗栓、抗炎效果。
✨ 科研启示
败血症不仅是免疫风暴,更是凝血风暴。本研究提醒我们,血小板除了止血,其作为“转录产物搬运工”和“免疫介质分泌者”的角色在危重症中可能更为关键。
✨ 临床/应用价值
HSP90α中和疗法可能成为败血症DIC的新曙光。与传统抗凝药或抗血小板药不同,这种“靶向胞外DAMP”的策略在理论上能抑制免疫损伤而不增加致命性出血风险,距离临床转化还需开展大规模成人及跨中心临床试验验证。
局限性
1、相对较小的样本队列可能无法充分捕捉败血症从早期到中晚期发展过程中血小板蛋白质组和细胞因子的动态变化;临床样本仅来源于儿童(平均年龄约4岁),考虑到儿童与成人在颗粒释放、整合素 αIIbβ3 表达及内化等方面的血小板功能差异,将结论直接外推至成人败血症时需保持谨慎
2、1G6-D7 中和抗体在临床转化中可能面临脱靶风险,因为 HSP90α 广泛表达,系统性抑制可能干扰健康细胞内蛋白质折叠和压力反应等必需的胞内伴侣功能,且 HSP90α 与 HSP90β 序列高度相似,可能引发非预期的免疫反应或对其他异构体产生影响
推荐阅读
Nature Communications | 瑞金医院团队揭示PHGDH介导的丝氨酸重编程:调控巨噬细胞炎症风暴的新机制
Science Immunology | 为什么女性疼痛更难缓解?密歇根州立大学研究发现雄激素调控单核细胞IL-10分泌,驱动疼痛缓解的性别差异
Nature Communications | 固有免疫传感器MDA5通过调控炎性衰老与蛋白质稳态延缓造血系统衰老

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 【郑州招教】郑州市第五十八中学2026年招聘教师公告(报名时间:即日起)
- 郑州高新区梧桐社区卫生服务中心招聘公告,大专起
- 郑州教师招聘,无笔试!郑州市英迪学校招聘教师公告【至2.28】
- 郑州的5元牛肉汤!
- 郑州市井风水密码!健康路/丰产路/陈寨,为啥这些夜市菜市场越逛越旺?
- 郑州轻工业大学关于启动2026届毕业生春季校园招聘活动的通知
- 郑州航空港南港的大型星级酒店项目
- 【3月19日郑州展】揭秘院士专家力荐的“战略单品”:GPC甘草多糖,25年专注呼吸道/结节!源头实力,厂家直招(展位号P121)
- 郑州颐和医院招聘学科带头人
- 河南郑州一店铺正月初九在派出所旁放“开门炮”被举报,派出所回应:涉事人员违规燃放烟花爆竹,被行政处罚
