磷脂酶PLPP1的缺失:
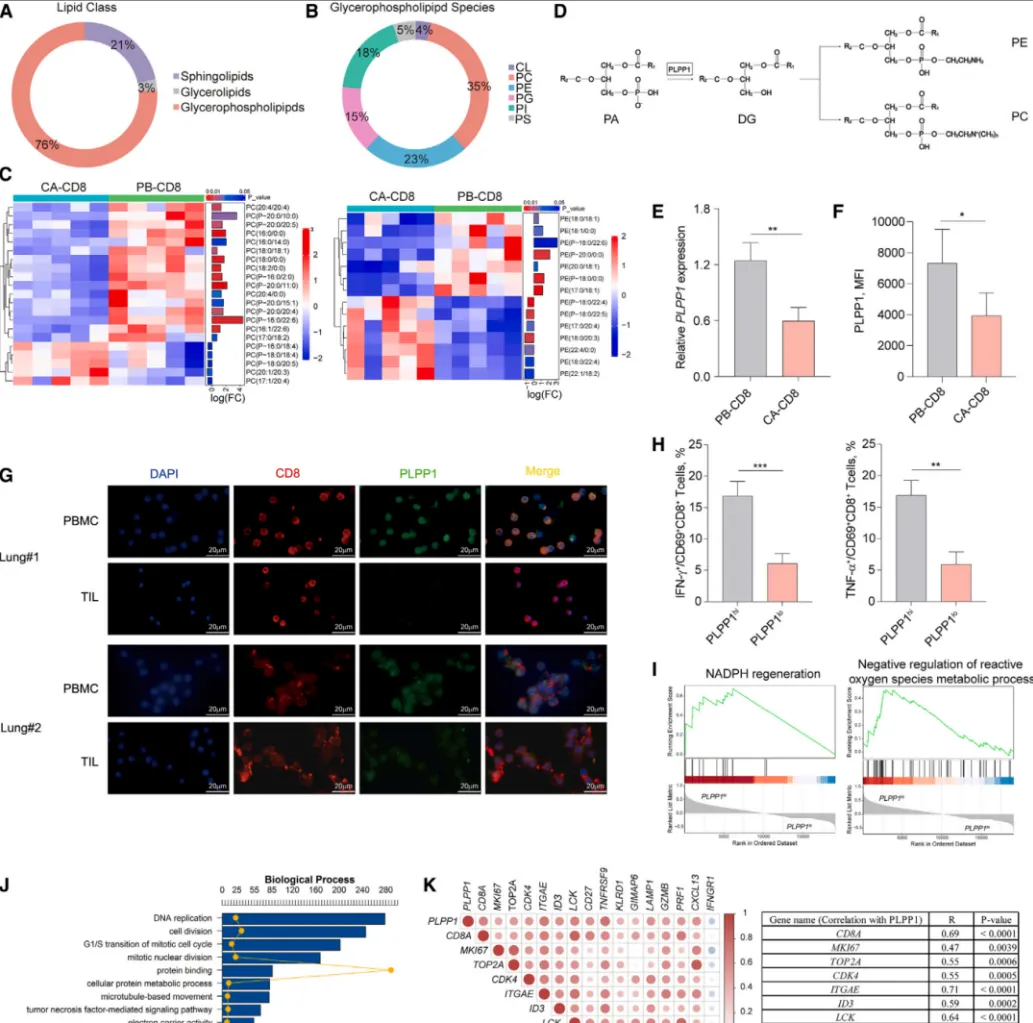
研究人员首先通过比较肺癌患者外周血与肿瘤组织中的CD8+ T细胞脂质谱,证实肿瘤内T细胞存在显著的磷脂代谢失调,具体表现为关键磷脂PC和PE的合成严重不足【图1A-C】。通过对合成通路的溯源分析,研究者将这一缺陷锁定在催化PC和PE合成的关键代谢酶——磷脂磷酸酶1(PLPP1)上【图1D】。
这一发现在肺癌、结肠癌及食管癌患者中得到了广泛验证:与循环中的T细胞相比,肿瘤浸润的CD8+ T细胞其PLPP1表达量均普遍显著下调【图1E-G】。功能关联分析显示,高表达PLPP1的CD8+ T细胞,其产生IFN-γ、TNF-α等关键效应细胞因子的能力更强,抗肿瘤功能更优【图1H】。生物信息学分析进一步表明,高PLPP1 T细胞富集了更强的增殖、活化及抗氧化应激相关基因特征【图1I, J】
 PLPP1缺失直接“触发”CD8+T细胞铁死亡:
PLPP1缺失直接“触发”CD8+T细胞铁死亡:
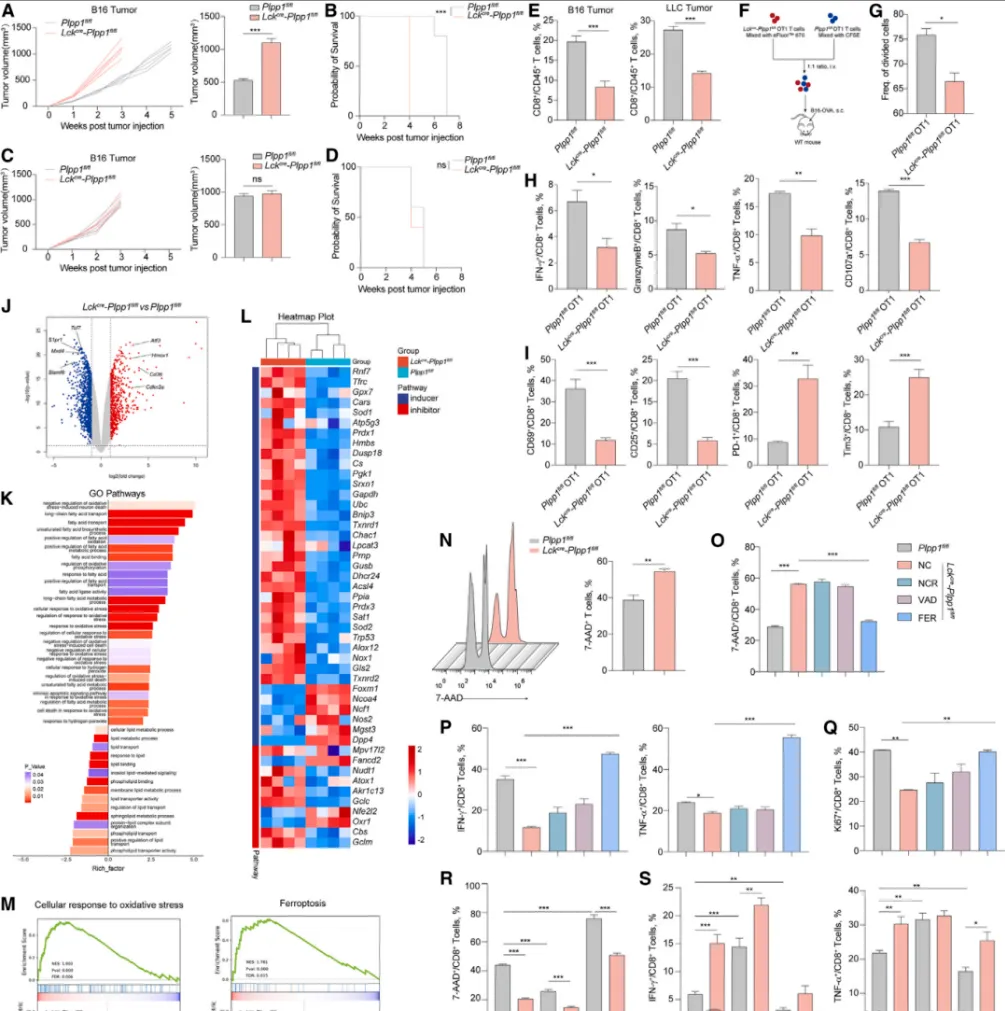
为明确PLPP1的功能,研究者构建了T细胞特异性敲除Plpp1的小鼠模型(Lckcre-Plpp1fl/fl)。实验证实,Plpp1敲除小鼠在接种黑色素瘤或肺癌细胞后,肿瘤生长显著加快,生存期明显缩短【图2A-D】。通过预先清除CD8+ T细胞进行反向验证,发现两组小鼠肿瘤生长无差异,确证表型源于CD8+ T细胞功能受损。
RNA-seq分析揭示,Plpp1缺失的CD8+ T细胞中,氧化应激与铁死亡相关通路基因显著富集【图2J-M】。细胞死亡实验进一步证实,Plpp1缺失导致的T细胞死亡可被铁死亡特异性抑制剂(Ferrostatin-1)挽救,而非凋亡或坏死抑制剂【图2N-Q】。一致性验证显示,铁死亡激动剂(RSL3)能模拟Plpp1缺失表型,而PlPP1过表达(Plpp1-OE)则能抵抗铁死亡【图2R-S】。
 脂肪酸加剧Plpp1缺失CD8+T细胞的铁死亡:
脂肪酸加剧Plpp1缺失CD8+T细胞的铁死亡:
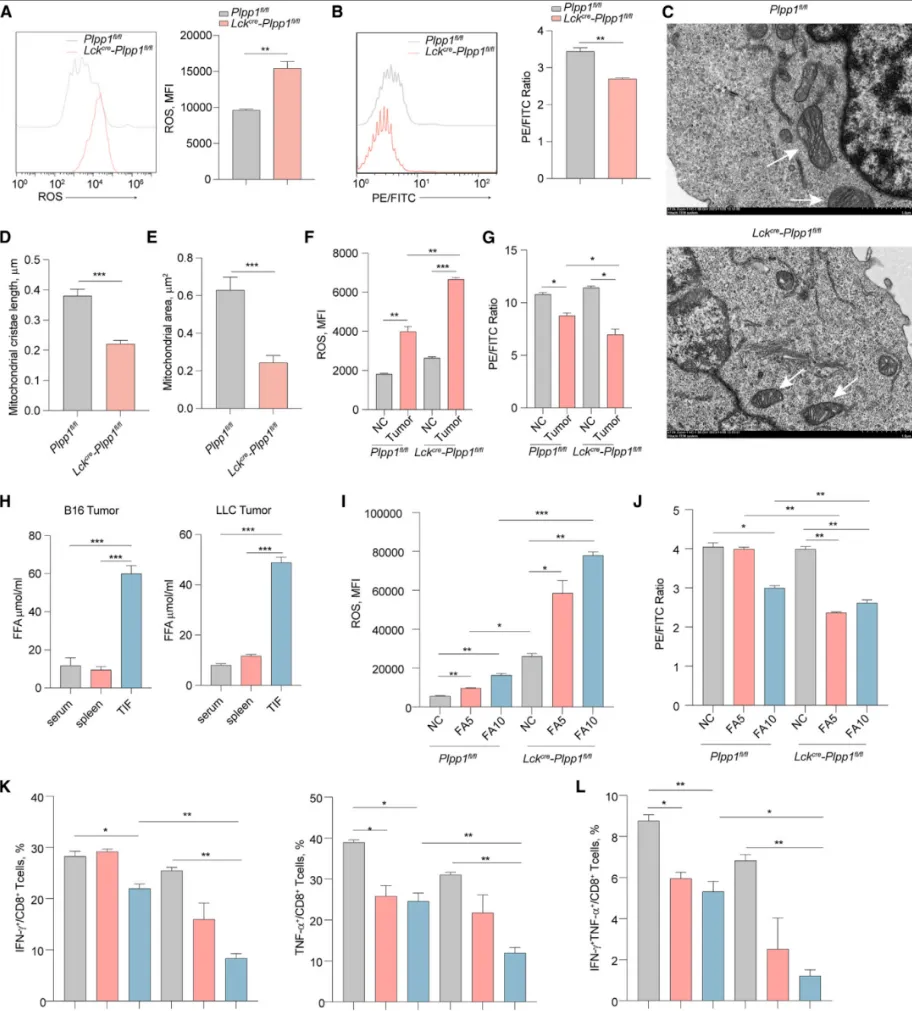
肿瘤微环境中的代谢物质协同促进了CD8+ T细胞的铁死亡。研究发现,Plpp1敲除的CD8+ T细胞内,ROS与脂质过氧化水平显著升高【图3A-B】,电镜下观察到了线粒体嵴紊乱等铁死亡典型超微结构改变【图3C-E】。
当这些细胞与肿瘤组织共培养后,ROS和脂质过氧化水平进一步急剧飙升【图3F-G】。分析发现,肿瘤间质液(TIF)中的游离脂肪酸含量远高于血清和脾脏【图3H】。
关键干预实验显示,用脂肪酸处理CD8+ T细胞,会显著加剧Plpp1敲除组的ROS/脂质过氧化水平,并更严重地损害其效应分子分泌功能【图3I-L】。这证明,肿瘤微环境中富集的游离脂肪酸,是特异性加剧Plpp1缺失T细胞铁死亡的关键“助推剂”。
 不饱和脂肪酸通过重塑磷脂种类诱导铁死亡:
不饱和脂肪酸通过重塑磷脂种类诱导铁死亡:
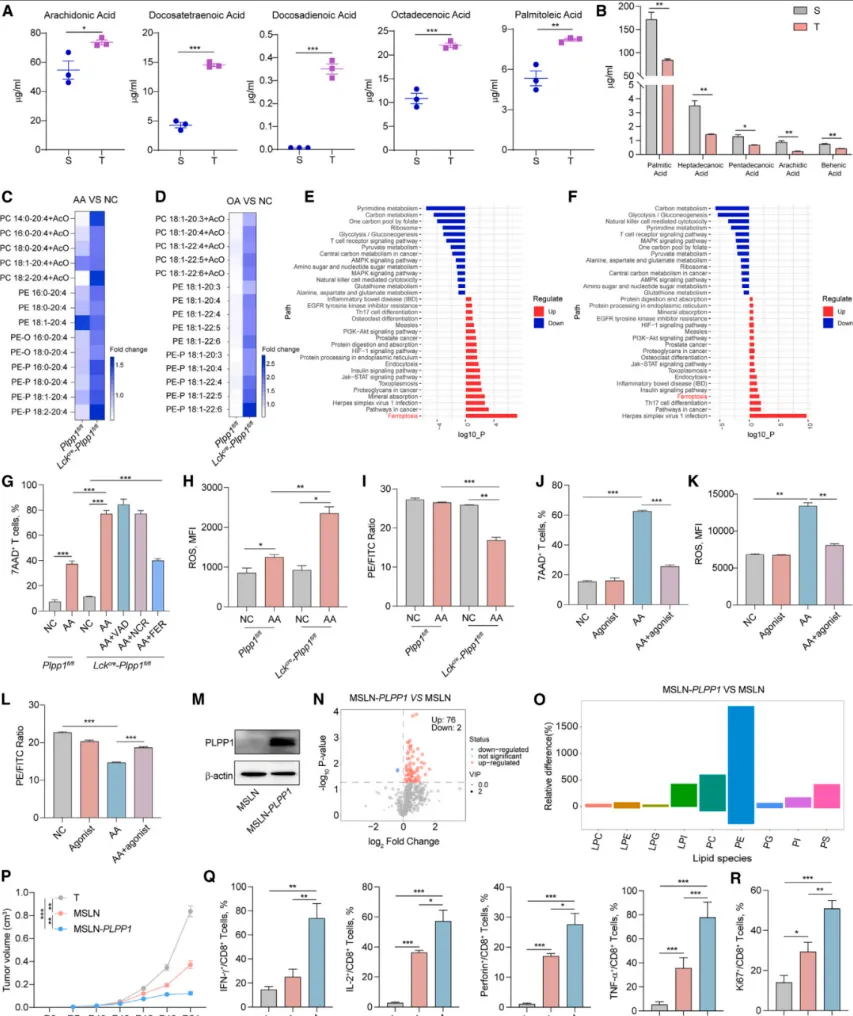
对肿瘤组织中脂肪酸种类的分析显示,不饱和脂肪酸(如花生四烯酸AA、油酸OA)含量显著升高,而饱和脂肪酸相对降低【图4A-B】。当Plpp1缺失的CD8+ T细胞暴露于AA或OA时,其活性氧水平、脂质过氧化程度和铁死亡率均显著高于对照细胞【图4C-D】。
这证实,肿瘤微环境中富集的不饱和脂肪酸是特异性触发Plpp1缺失CD8+ T细胞铁死亡的关键环境因素。作为对照,实验进一步发现,过表达PLPP1的CAR-T细胞展现出更强的磷脂水平、抗肿瘤活性和体内外增殖能力【图4E-R】。
 体外实验:体外验证实验发现,当CD8+ T细胞与肿瘤细胞共培养时,其PLPP1的表达水平会显著下降。而使用PD-1的特异性阻断抗体(而非CTLA-4阻断抗体)可以逆转这一效应,有效恢复CD8+ T细胞中PLPP1的表达【图5A-C】。这直接证明了PD-1信号通路是肿瘤微环境中负向调控CD8+ T细胞PLPP1表达的上游关键信号。
体外实验:体外验证实验发现,当CD8+ T细胞与肿瘤细胞共培养时,其PLPP1的表达水平会显著下降。而使用PD-1的特异性阻断抗体(而非CTLA-4阻断抗体)可以逆转这一效应,有效恢复CD8+ T细胞中PLPP1的表达【图5A-C】。这直接证明了PD-1信号通路是肿瘤微环境中负向调控CD8+ T细胞PLPP1表达的上游关键信号。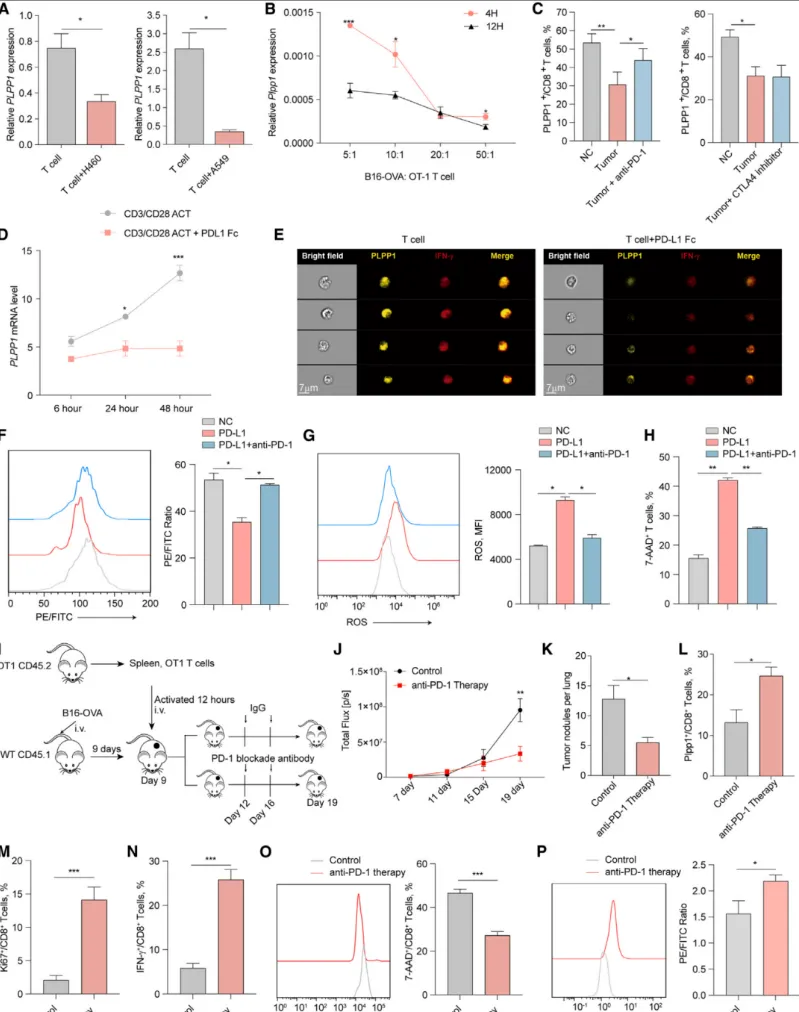 信号通路解析:
信号通路解析:
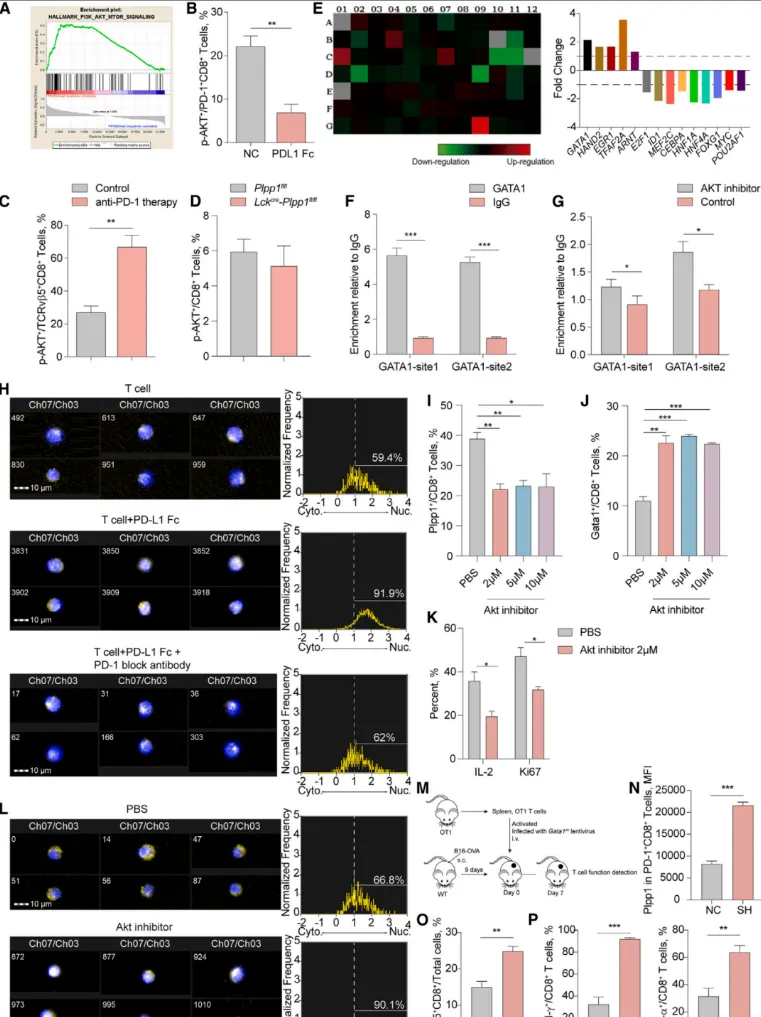
机制研究发现,激活PD-1信号会抑制下游的Akt通路。通过转录因子筛选和染色质免疫共沉淀实验证实,Akt抑制会促进转录因子GATA1入核,并直接结合到PLPP1基因的启动子区域,从而抑制PLPP1的转录表达【图6A-H】。
功能回复实验进一步证明,人为敲低GATA1后,即使PD-1信号被激活,PLPP1的表达仍能维持,且T细胞的抗肿瘤功能随之显著增强【图6I-P】。这确定了GATA1是连接PD-1信号与PLPP1表达抑制的关键分子中介。
 临床意义:PLPP1水平决定免疫治疗疗效:
临床意义:PLPP1水平决定免疫治疗疗效:
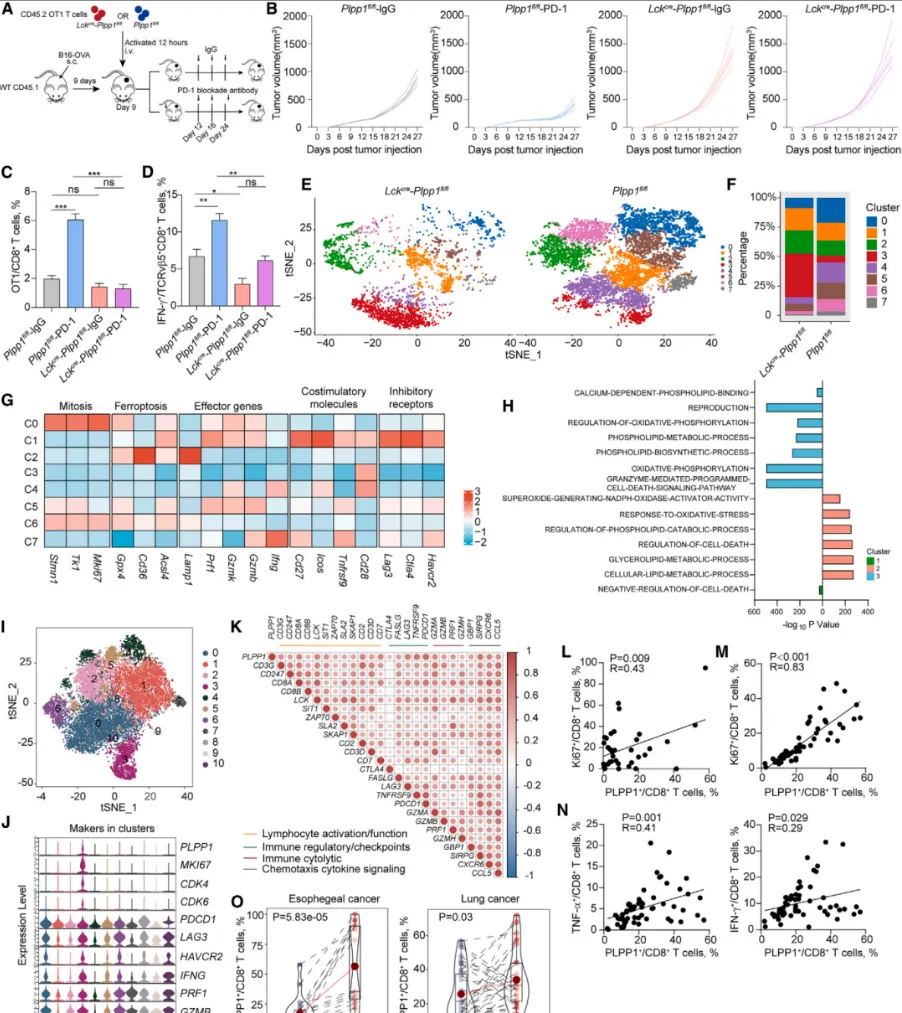
在荷瘤小鼠模型中,PD-1抑制剂能够显著提升肿瘤内T细胞的PLPP1表达,并降低其铁死亡水平,从而发挥抗肿瘤疗效。然而,对于T细胞特异性敲除Plpp1的小鼠,PD-1抑制剂则完全失去治疗作用【图7A-D】。这证明,PD-1抑制剂发挥疗效至少部分依赖于其对PLPP1表达与功能的恢复。
对临床样本的分析印证了这一发现:在食管癌和肺癌患者中,CD8+ T细胞的PLPP1表达量与细胞增殖标志物Ki67及效应因子表达呈正相关。更为关键的是,在接受抗PD-1治疗的患者中,治疗后其T细胞的PLPP1表达量显著上升【图7E-O】,为机制的临床相关性提供了直接证据。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
