郑州大学肖廷辉&沈若凡Angew:氢溢流加速 + 等离子体效应!磷酸根 MXene 调控 Ru-Cu 单原子氨硼烷水解制氢
绿色氢能是化石燃料的重要替代能源,氨硼烷(AB)因储氢量高、结构简单且稳定性好,成为水解制氢的理想储氢材料,其水解制氢的决速步为水离解。单原子合金(SAA)因独特电子结构可降低反应能垒,是高效催化剂设计方向,而氢溢流效应对调控水离解动力学至关重要。MXene作为催化剂载体虽能修饰催化剂电子结构,但其直接参与水离解催化过程、加速反应动力学的潜力尚未被开发。
氨硼烷水解制氢的水离解步骤能垒较高,成为反应速率的关键限制因素,亟需优化催化剂以降低该步骤能垒。
传统MXene基催化剂载体仅能通过电子修饰间接提升催化活性,其直接与反应物/产物相互作用、加速催化动力学的功能未被挖掘。
单原子合金催化剂的催化性能提升仅聚焦于活性位点设计,缺乏对载体的精准调控,未实现载体-催化剂-反应物的协同作用。
光辅助催化制氢中,如何有效利用等离子体效应激发热电子、进一步活化反应分子并降低反应能垒,仍需探索可行的催化剂设计策略。
成功制备磷酸根阴离子工程化MXene(P-Ti₃C₂)负载的Ru-Cu单原子合金催化剂,在室温光照条件下实现746 L g_Ru⁻¹ min⁻¹的高质量比活性,远超现有Ru-Cu基制氢催化剂。
磷酸根阴离子可调控MXene表面Ti位点的电子结构,显著加速氢溢流效应,同时Ru-Cu单原子合金的集合效应能有效降低O-H键断裂能垒。
Ru-Cu单原子合金存在局域表面等离子体共振(LSPR)效应,光照下激发的热电子可进一步活化氨硼烷和水分子,使水离解能垒从0.83 eV降至0.63 eV。
该催化剂具有优异的循环稳定性,经9次连续循环后仍保留92.6%的初始活性,且活性金属无明显团聚、流失,电子结构保持稳定。
催化剂中Ru-Cu以原子级分散形成合金,通过Ti-O-Ru键锚定于P-Ti₃C₂载体,实现了载体-催化剂间的高效电子转移,优化了催化界面电荷分布。
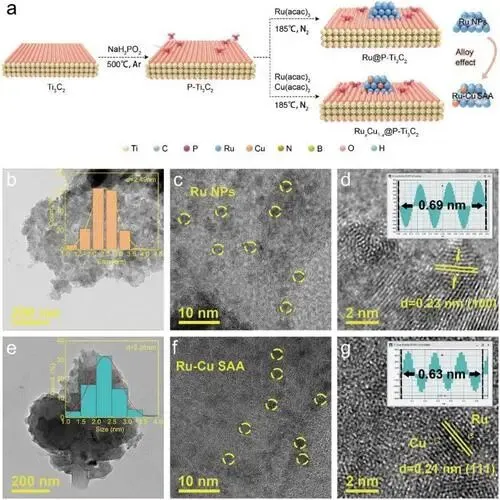
合成流程:先通过高温煅烧将次磷酸钠引入Ti₃C₂表面得到P-Ti₃C₂,再将Ru、Cu前驱体经乙二醇溶解后与P-Ti₃C₂混合,高温还原得到RuₓCu₁₋ₓ@P-Ti₃C₂系列催化剂,Ru@P-Ti₃C₂则为无Cu的对照样。
Ru@P-Ti₃C₂形貌:Ru纳米颗粒呈层状分布,存在轻微团聚,平均粒径2.49 nm,高分辨TEM显示0.23 nm晶格条纹对应金属Ru的(100)晶面。
Ru₉Cu₁@P-Ti₃C₂形貌:Ru-Cu单原子合金分散性更优,平均粒径减小至2.28 nm,0.21 nm晶格条纹对应Ru-Cu SAA结构;无明显Cu晶格条纹,证明Cu原子以原子级均匀分散在Ru晶格中。
粒径减小提升了催化剂的比表面积和原子利用率,为催化反应提供更多活性位点。
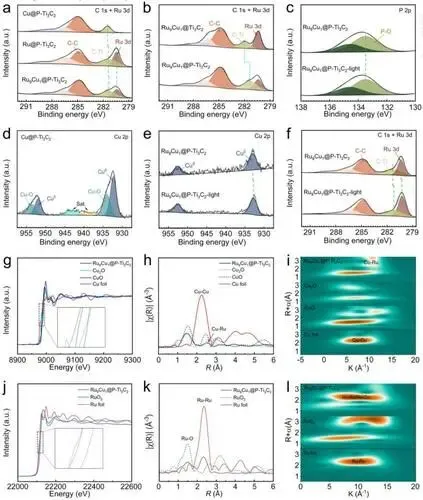
XPS表征:C 1s和Ru 3d谱中280.44 eV峰对应金属Ru⁰,P 2p谱133.5 eV峰证实P⁵+成功引入;Ru₉Cu₁@P-Ti₃C₂的C-Ti峰负移,表明磷酸根为Ti位点提供电子,Cu 2p峰正移证明Cu向Ti转移电子;光照下Ti、Ru、Cu位点均出现电子累积,为等离子体热电子转移所致。
XANES表征:Cu K边和Ru K边吸收边与金属箔一致,证实Ru、Cu均以金属态存在于催化剂中。
EXAFS及WT-EXAFS表征:Cu K边在2.51 Å处出现Cu-Ru配位峰,无Cu-Cu峰,Ru K边观察到Ru-Ru键长收缩,且存在Ru-Cu配位;催化剂通过Ti-O-Ru键锚定载体,无Ru-Ti直接配位,证实Ru-Cu SAA的成功合成。
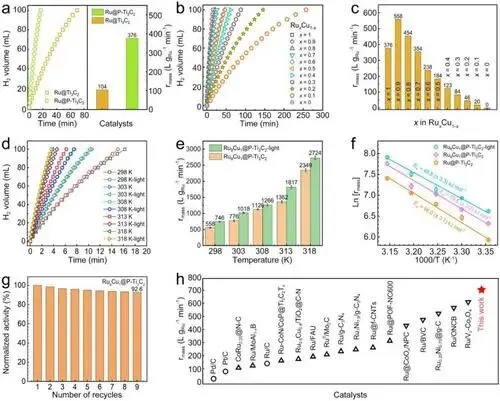
磷酸根的作用:Ru@P-Ti₃C₂的制氢速率是Ru@Ti₃C₂的近4倍,证明磷酸根对Ti₃C₂的电子调控可显著提升催化活性。
合金比例优化:RuₓCu₁₋ₓ@P-Ti₃C₂中Ru₉Cu₁比例表现出最优催化性能,暗态下质量比活性达558 L g_Ru⁻¹ min⁻¹,归因于Ru-Cu SAA的集合效应。
光照的提升效果:室温光照下Ru₉Cu₁@P-Ti₃C₂的质量比活性提升至746 L g_Ru⁻¹ min⁻¹,表观活化能从暗态的53.6 kJ mol⁻¹降至49.8 kJ mol⁻¹。
稳定性测试:9次循环后催化剂保留92.6%初始活性,活性金属无团聚、流失,电子结构稳定。
性能对比:该催化剂的质量比活性显著优于商用催化剂及已报道的Ru基氨硼烷水解催化剂。
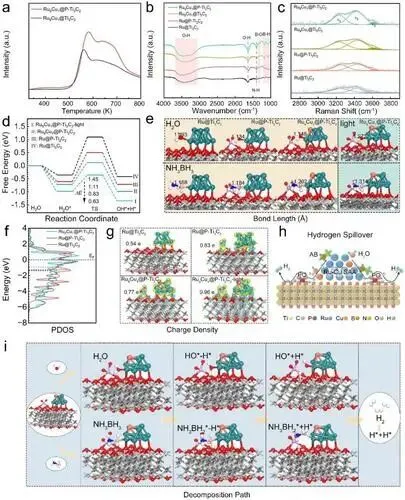
氢溢流表征:H₂-TPD结果显示Ru₉Cu₁@P-Ti₃C₂的高温溢流峰面积更大,证明磷酸根工程化可显著提升氢溢流效率;H₂-D₂交换实验表明其氢物种的解离、迁移和复合效率更高。
原位谱学表征:原位FT-IR显示Ru₉Cu₁@P-Ti₃C₂的B-H键特征峰最强,水吸附和AB水解活性最高;原位拉曼显示其表面自由水(易水解)比例更高,水离解能力更强。
DFT理论计算:Ru₉Cu₁@P-Ti₃C₂的水离解能垒为0.83 eV,远低于Ru@P-Ti₃C₂(1.11 eV)和Ru@Ti₃C₂(1.45 eV),光照下能垒进一步降至0.63 eV;O-H和B-H键在该催化剂表面发生显著伸长,分子活化程度更高。
电子结构分析:Ru₉Cu₁@P-Ti₃C₂中Ru的d带中心更靠近费米能级,与O-H键反键轨道相互作用更强;电荷密度差显示磷酸根、Ru-Cu合金及光照可逐步提升催化剂表面电荷累积,优化电子转移。
催化机理:AB和H₂O先吸附于Ru位点,Ru/Ru-Cu位点活化并断裂B-H/O-H键,氢原子通过氢溢流迁移至P-Ti₃C₂表面并复合为H₂脱附,Cu作为助催化剂结合光照热电子进一步提升分子活化效率。
原文链接:https://onlinelibrary.wiley.com/doi/10.1002/anie.202524246




 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
