研究背景
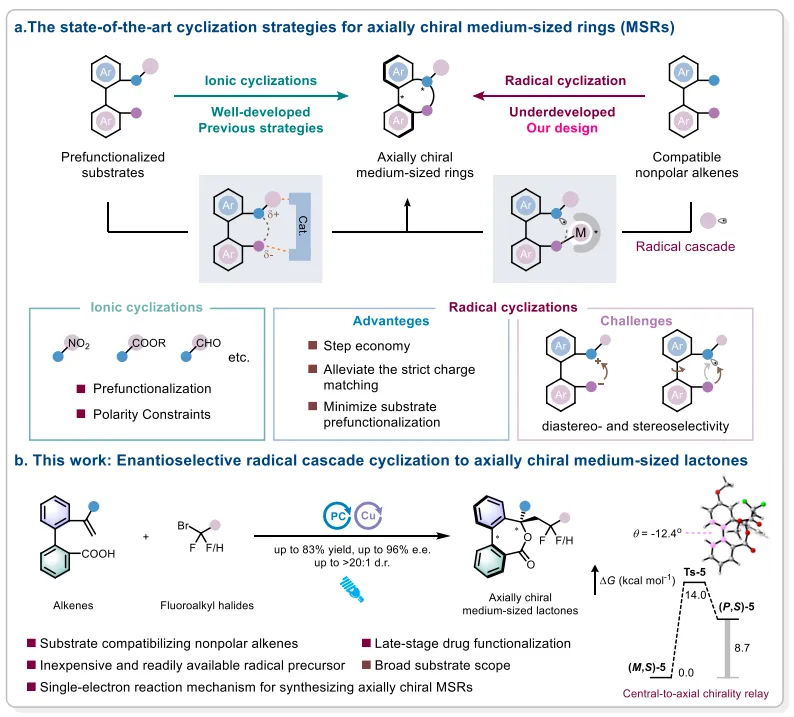
轴手性中环骨架(MSRs)因其兼具结构刚性与构象柔性,在药物化学、材料科学和不对称催化等领域具有重要应用价值。现有催化不对称合成策略主要依赖预先构建的环状底物(如动力学拆分和合成后修饰),限制了结构多样性;而直接环化方法大多基于离子型路径,需底物预先官能团化以满足严格电子匹配要求。近年来,自由基级联环化在构建五、六元环方面取得显著进展,但将其应用于轴手性MSRs仍面临挑战:中环关环缓慢且熵不利,与快速进行的自由基加成存在动力学不匹配,压缩了催化剂控制立体选择性的时间窗口,且氧化条件下易发生非选择性背景离子环化。因此,发展无需底物预官能团化、兼容非极化烯烃的自由基级联策略构建轴手性MSRs具有重要意义。近日,郑州大学贾师琦、蓝宇团队在Angewandte Chemie International Edition期刊上发表了题为“Enantioselective Radical Cascade Cyclization to Axially Chiral Medium-Sized Lactones”的论文。开发了一种协同光氧化还原/手性铜催化的不对称自由基级联环化反应,以非极化烯烃和氟烷基卤化物为原料,通过单电子转移(SET)生成烷基自由基,随后对烯烃进行自由基加成并通过C–O键形成完成七元环内酯的构建。手性铜催化剂通过羧酸根配位与底物结合,建立了明确且构象偏置的手性环境,实现了对自由基捕获和环化过程的高效立体控制。该反应以高达83%的产率、96%的对映选择性和>20:1的非对映选择性获得轴手性中环内酯产物。研究还通过生物活性分子的后期官能团化展示了合成实用性,并结合理论计算揭示了立体决定步骤发生在C–O键形成阶段,热力学驱动的中心到轴手性传递机制确保了七元环中轴手性的稳定。
突破离子型路径的底物限制:采用自由基级联策略,以商业可得的烷基卤化物为自由基前体,无需底物预先官能团化,兼容非极化烯烃,有效规避了传统离子环化对严格电子匹配和极性约束的依赖。
高立体控制效率:通过羧酸根配位的手性铜配合物建立构象偏置的手性口袋,实现了高达96% e.e.和>20:1 d.r.的优异立体选择性,所有产物均为单一非对映异构体。
阐明立体决定机制:理论计算与IGMH分析表明,立体决定步骤为自由基取代介导的C–O键形成过程,而非传统的氧化加成路径; favored过渡态中配体与二氟甲基取代基之间的空间排斥(H–H距离2.0 Å)是导致选择性差异的关键因素。
热力学驱动的中心到轴手性传递:尽管七元环通常具有较低的轴翻转能垒,本研究通过计算揭示了热力学驱动的中心到轴手性传递机制(ΔG‡ = 1.1 kcal/mol),该机制通过锁定构象将中心手性信息传递至轴手性,从而稳定了中环骨架的轴手性。
广泛的底物适用性与合成实用性:反应兼容多种氟烷基卤化物(甲基、乙基、苄基酯等)、酰胺底物以及不同取代模式的烯烃;成功实现了天然产物(如薄荷醇、葑醇、α-萜品醇)和药物分子(如苯佐卡因、胆固醇衍生物)的后期官能团化,并完成了克级规模合成(2.846 g产物,95% e.e.)。
该研究通过协同光氧化还原与手性铜催化,成功实现了不对称自由基级联环化反应,为轴手性中环内酯的构建提供了一种高效、通用的新策略。该方法以商业可得的氟烷基卤化物和非极化烯烃为原料,突破了传统离子型环化对底物预官能团化和严格电子匹配的依赖,在温和条件下以优异的产率和对映选择性(up to 83% yield, 96% e.e., >20:1 d.r.)构建了含多重手性元素的中环骨架。机理研究表明,C–O键形成步骤为立体决定步骤,且热力学驱动的中心到轴手性传递机制有效稳定了七元环的轴手性。该工作不仅拓展了自由基化学在轴手性中环合成中的应用边界,也为相关生物活性分子的后期修饰提供了新工具。未来研究可进一步拓展该策略至八元及更大环系、探索非氟烷基自由基前体的适用性,并结合连续流光化学等技术提升反应效率,推动轴手性中环化合物在药物发现和材料科学中的实际应用。
Y. Hao, J. Chen, N. Hu, et. al., Enantioselective Radical Cascade Cyclization to Axially Chiral Medium-Sized Lactones, Angew. Chem. Int. Ed., 2026, e3596438.
链接:https://doi.org/10.1002/anie.3596438以上内容均为小编个人观点,仅供参考,如有不当之处,敬请批评指正!
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
