 点击蓝字,关注我们,每天有干货
点击蓝字,关注我们,每天有干货
第一作者:李雯雯、胡志昂、于镜坤
通讯作者:卢思宇、常江伟
通讯单位:郑州大学
论文DOI:10.1002/anie.2263598
海水电解是直接利用丰富海水资源生产绿氢、解决淡水资源约束的理想途径,但其实际应用面临高浓度氯离子的严重腐蚀这一根本性挑战。在阳极,Cl⁻会与析氧反应竞争吸附,不仅诱发化学腐蚀,更会导致有害的析氯副反应,生成侵蚀性物种,快速降解催化剂并破坏电极。传统物理屏障或界面修饰等抗腐蚀策略,常因阻碍反应物传输或在高电流下失效,难以在高活性、高选择性、强抗腐蚀性三者间取得平衡,导致系统寿命普遍较短(<500小时),成为制约海水电解技术发展的核心瓶颈。
本研究创新性地提出一种“碳化聚合物点界面工程”策略,成功设计出兼具超高活性和非凡抗氯腐蚀能力的Co₃O₄@CPDs阳极催化剂。其核心亮点在于:1) 创纪录的活性和超长稳定性:在模拟碱性海水中,该催化剂在500 mA cm⁻²工业电流密度下过电位仅为270 mV,并在600 和 800 mA cm⁻²下分别实现了4200和3200小时的惊人稳定运行。2) 揭示动态、智能的双重保护机制:CPDs不仅稳定催化剂晶格氧、优化反应路径以提升本征活性,其自身在反应中被电氧化生成的CO₃²⁻物种,更在表面动态形成了一层能选择性阻挡Cl⁻吸附与穿透,却允许OH⁻自由传输的“智能”保护层。3) 验证实用化器件性能:基于该催化剂的阴离子交换膜模拟海水电解槽在1.0 A cm⁻²、60°C下稳定运行超过1500小时,电池电压仅1.73 V,展现了巨大的实际应用潜力。
本研究旨在攻克海水电解阳极催化剂易受氯离子腐蚀失活的核心难题。研究团队设计并制备了一种新型复合催化剂:将碳化聚合物点锚定在尖晶石Co₃O₄表面,构建了Co₃O₄@CPDs异质结构。
该设计的精妙之处在于其协同作用机制。首先,CPDs对Co₃O₄本征活性的提升:理论计算与实验表明,CPDs的引入稳定了Co₃O₄中的晶格氧,降低了反应能垒,促使氧析出反应机制从易导致结构重构的晶格氧机制,向更温和、高效的吸附质演化机制转变,从而提升了催化本征活性与稳定性。
其次,也是更关键的,CPDs构建了动态、选择性的抗腐蚀界面。在电解过程中,锚定在表面的CPDs被部分电化学氧化,原位生成CO₃²⁻ 物种。这些CO₃²⁻与CPDs残基共同在催化剂表面形成一层稳定的界面层。该层的关键功能在于其“离子选择性”:它能够有效排斥和阻挡体积较大、吸附性强的Cl⁻离子,防止其接触并毒化活性位点;同时,其对体积小、迁移快的OH⁻离子阻力很小,保证了反应物OH⁻的快速供应。这种“拒Cl⁻而迎OH⁻”的智能特性,完美解决了传统保护层“一堵了之”导致的传质受限问题。
性能验证表明,该催化剂在模拟海水电解中实现了工业级电流密度下的超高活性和超长寿命。组装的阴离子交换膜电解槽也展现了卓越的长期运行稳定性与低能耗。该工作通过“CPDs优化本征活性 + 原位生成智能动态保护层”的双重策略,为设计下一代适用于苛刻真实环境的高性能、高稳定性电解水催化剂提供了全新的范式。
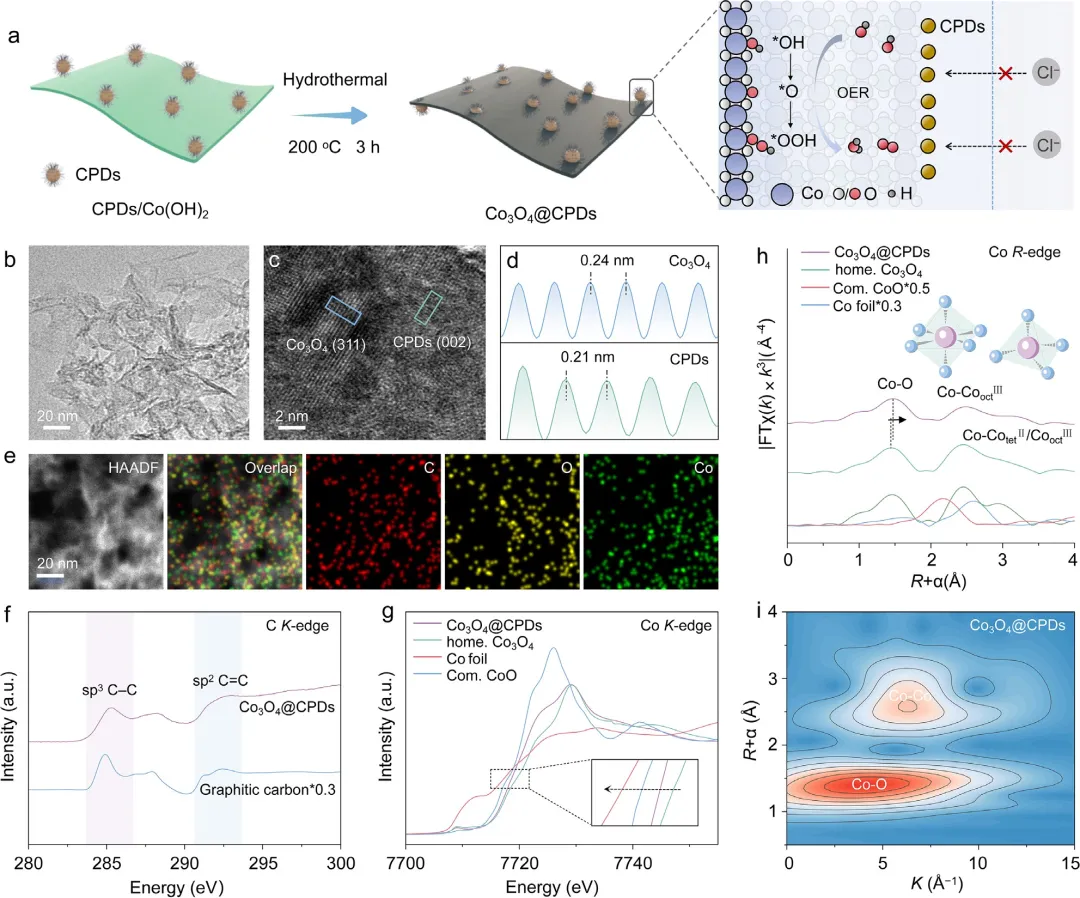
图1 | Co₃O₄@CPDs的合成与结构表征。(a) Co₃O₄@CPDs的合成过程示意图。(b) HAADF-STEM揭示的Co₃O₄@CPDs的纳米片形貌。(c) Co₃O₄@CPDs的高分辨率HAADF-STEM图像(上方)及对应的放大图(下方)。(d) 图(c)中蓝色和绿色虚线区域的线扫描强度分布图。(e) Co₃O₄@CPDs中C、Co和O的EDS元素分布图。(f) Co₃O₄@CPDs与石墨粉的C K-edge XANES对比。(g) Co₃O₄@CPDs、Co箔、商品CoO和自制Co₃O₄的归一化Co K-edge XANES谱图。插图示出Co₃O₄@CPDs中Co的降低价态。(h) Co₃O₄@CPDs、自制Co₃O₄、商品CoO和Co箔的FT Co K-edge EXAFS谱图。(i) Co₃O₄@CPDs的k³加权WT-EXAFS等高线图。
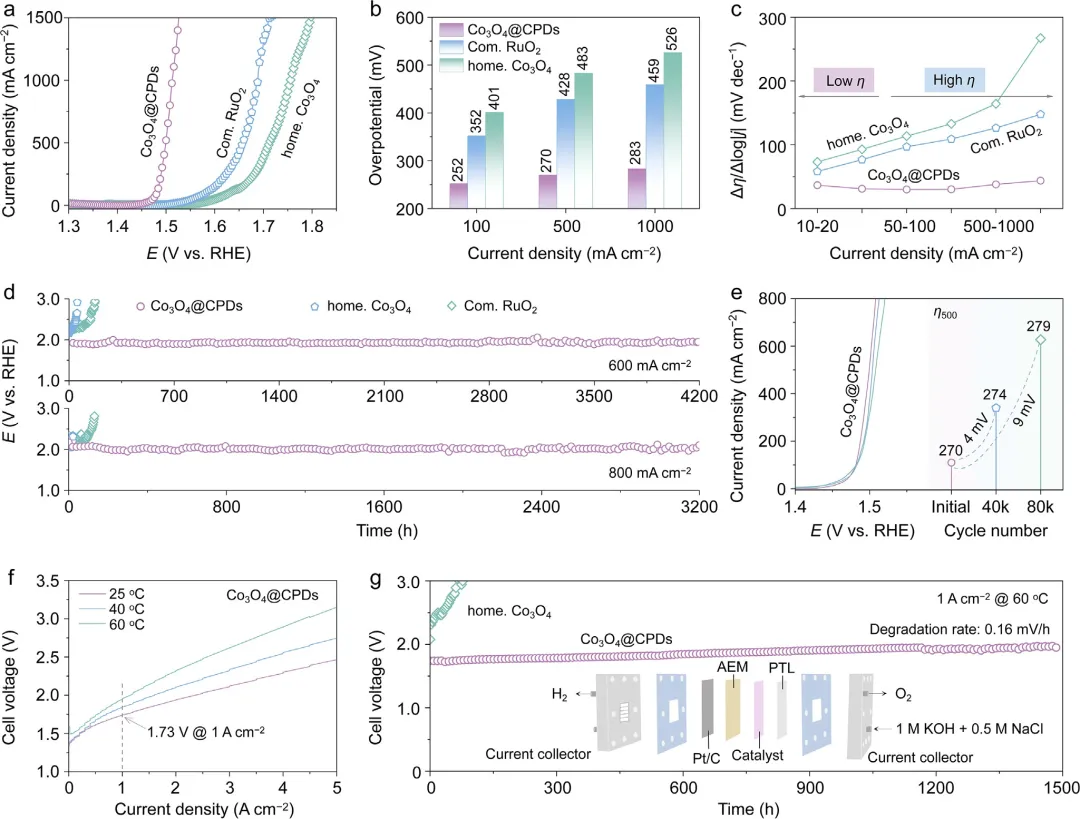
图2 | OER性能。(a) Co₃O₄@CPDs、自制Co₃O₄和商品RuO₂在1.0 M KOH + 0.5 M NaCl中的LSV曲线。(b) 在j₁₀₀、j₅₀₀和j₁₀₀₀处的对应过电位。(c) 不同电流密度下电催化剂的Δη/Δlog|j|值。(d) 不同电催化剂在j₆₀₀和j₈₀₀下的计时电位曲线(无iR补偿)。(e) 加速电压循环测试前后的稳态OER极化曲线对比。(f) Co₃O₄@CPDs在AEMSE中记录的极化曲线。(g) 基于Co₃O₄@CPDs的电解槽在1.0 A cm⁻²下的稳定性。电解槽在60℃下运行,所有AEMSE的电压均未进行iR补偿。
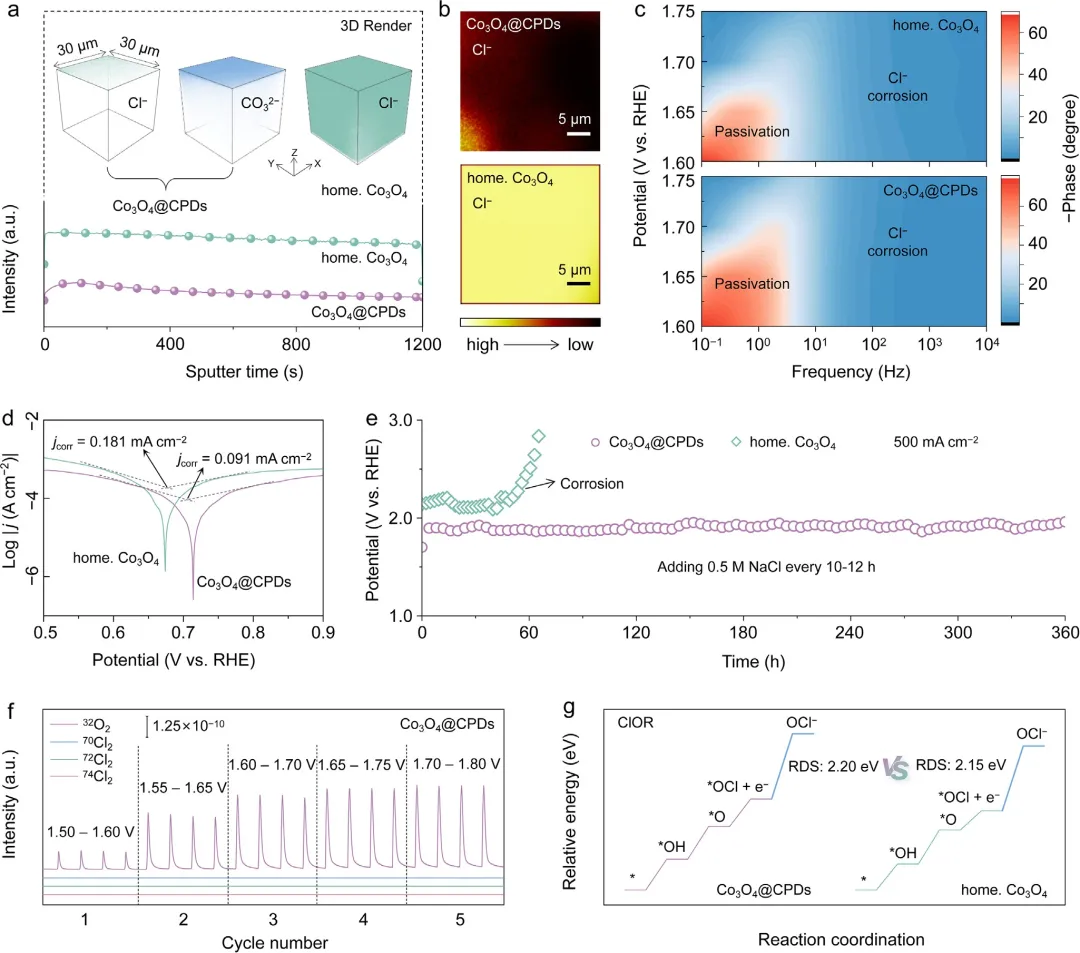
图3 | Co₃O₄@CPDs抗Cl⁻腐蚀性能评估。(a) 在j₅₀₀稳定性测试后,Co₃O₄@CPDs和自制Co₃O₄中Cl物种的TOF-SIMS深度分布图。插图为TOF-SIMS负离子深度分布三维图。(b) TOF-SIMS负离子分布图。(c) Co₃O₄@CPDs和自制Co₃O₄在相同NaCl浓度下的原位EIS谱。(d) Co₃O₄@CPDs和自制Co₃O₄的腐蚀电位测试。(e) Co₃O₄@CPDs和自制Co₃O₄在j₅₀₀下的计时电位响应(初始测试在含0.5 M NaCl的1.0 M KOH中进行,每10-12小时补充0.5 M NaCl)。(f) Co₃O₄@CPDs在1.0 M KOH + 0.5 M NaCl中的原位DEMS图。(g) ClOR在Co₃O₄@CPDs和自制Co₃O₄上的吉布斯自由能图。
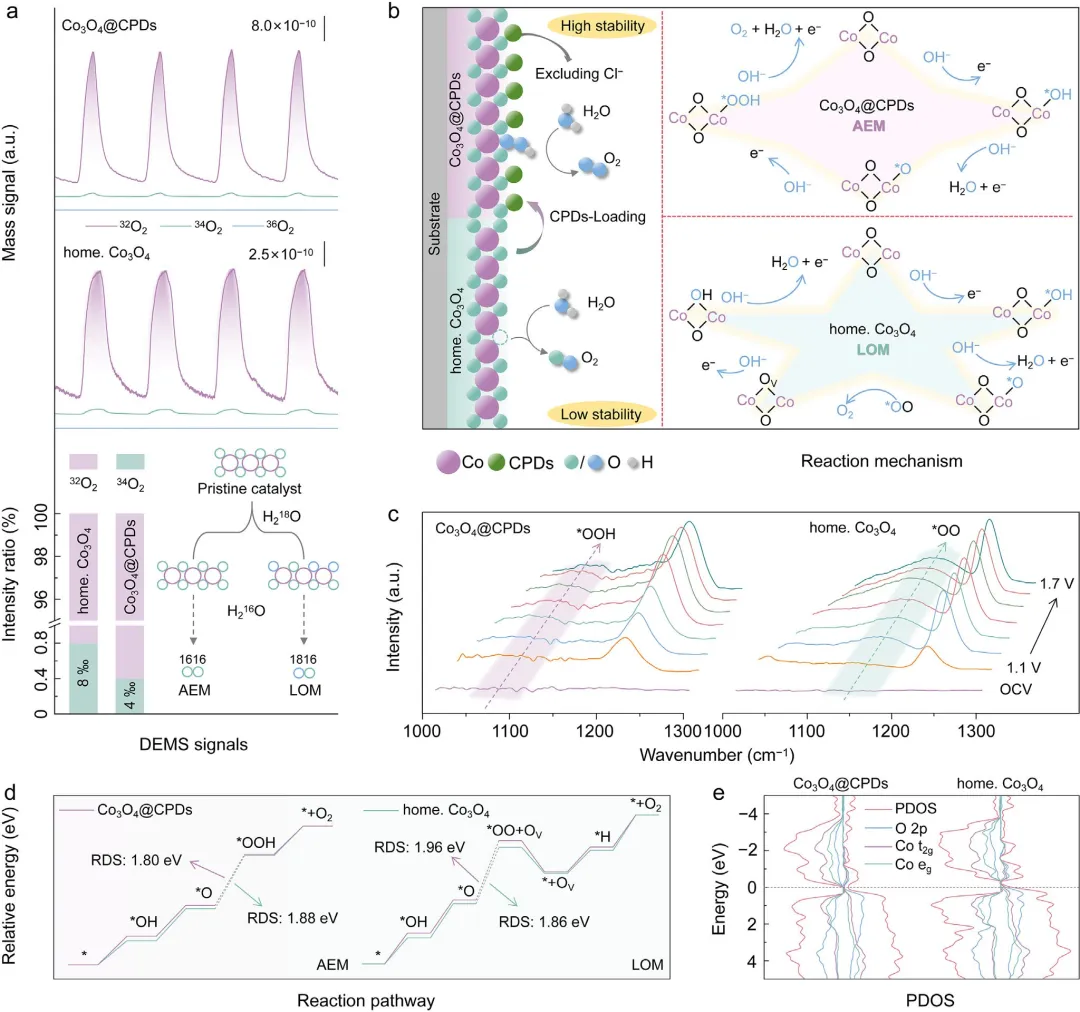
图4 | 原位机理研究与DFT计算。(a) 在H₂¹⁶O电解液中对¹⁸O标记的Co₃O₄@CPDs(上方)和¹⁸O标记的自制Co₃O₄(中间)进行LSV测试期间,通过原位DEMS采集的³²O₂、³⁴O₂和³⁶O₂信号。下图详细比较了³²O₂、³⁴O₂和³⁶O₂信号。插图为DEMS区分AEM和LOM路径的示意图。(b) Co₃O₄@CPDs上的AEM路径(上方)和自制Co₃O₄上的LOM路径(下方)。(c) Co₃O₄@CPDs(左)和自制Co₃O₄(右)上OER过程的原位ATR-SEIRAS研究。(d) Co₃O₄@CPDs和自制Co₃O₄上遵循AEM和LOM路径的ΔG图对比。(e) Co₃O₄@CPDs和自制Co₃O₄的PDOS图,虚线表示费米能级。
该研究开发了一种Co₃O₄@CPDs电催化剂,能够在工业电流密度下实现高效、选择性和持久的碱性海水电解。该催化剂在1500 mA cm⁻²的析氧反应中仅需296 mV过电位,并在600 mA cm⁻²下维持了超过4200小时的稳健长期运行。在AEMSE中,该催化剂在1.0 A cm⁻²和1.73 V的竞争性电池电压下,展现出超过1500小时的使用寿命。原位表征和DFT模拟揭示,表面锚定的CPDs通过静电排斥作用排斥Cl⁻,同时原位形成的CO₃²⁻协同构成了多层保护屏障,保护阳极免受腐蚀性电解液的攻击。此外,CPDs调控了Co₃O₄的电子结构并抑制了晶格氧的参与,确保了即使在高Cl⁻浓度和大电流密度下,AEM在整个OER电位范围内仍占主导地位,从而同时提升了活性和稳定性。该研究为海水电解提供了一种实用的高性能催化剂,并为设计稳健、耐氯的电催化剂提供了基本原理。
卢思宇,男,1986年生,郑州大学化学学院教授、博士生导师,国家优秀青年科学基金获得者,现任郑州大学碳点研究中心主任。2005年至2016年于吉林大学完成本科、硕士及博士学业,2016年起任职于郑州大学 [2-4]。主要从事碳点等光电纳米晶及其复合材料的可控构筑与光电性质研究,在《Advanced Materials》《Angewandte Chemie International Edition》等期刊发表SCI论文200余篇,总被引24000余次(Google Scholar),H因子91,60篇入选ESI高被引论文。主持国家自然科学基金优秀青年科学基金项目、面上项目及青年项目。入选2022年度科睿唯安全球高被引科学家及斯坦福大学全球前2%顶尖科学家榜单。担任《Chinese Chemical Letters》编委及《Science China Chemistry》《Energy & Environmental Materials》等期刊青年编委,培养研究生14名,其中6人次获国家奖学金。
常江伟,研究员,博士生导师。河南省高层次人才,中原英才青年拔尖人才,河南省优青。2021年于大连理工大学获博士学位,师从邱介山教授和于畅教授。致力于新结构高性能碳材料的可控构筑及高性能电池、能源催化应用研究,至今以第一或通讯作者在Nat. Synth., Nat. Commun.,J. Am. Chem. Soc., Angew. Chem. Int. Ed., Matter等能源期刊发表论文近30余篇,5篇论文入选ESI前0.1%热点文章,7篇文章入选ESI前1%高被引论文,1篇研究成果在国家自然科学基金会官方专题报道。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
